Output File Formats in MD++
This tutorial describes the formats of MD++ output files. The most frequently used file in MD++ is the configuration (.cn) file describing atomic positions (and other attributes). Also used a lot is the property (.out) file produced by Molecular Dynamics simulations. MD++ can also read and write VASP (POSCAR) and LAMMPS files.
Contents |
Configuration .cn File
In MD++, we save the current atomic configuration (in memory) to a file using the following command.
finalcnfile = myfile.cn writecn
(This is what we write in a .script input file. If we use .tcl input file, we need to put MD++ at the beginning of the line.)
The atomic configuration can be read into MD++ later (either in the same simulation run or in a different run) using the following command.
incnfile = myfile.cn readcn
Different amount of details are saved into the file, depending on two flags: writevelocity and writeall. By default, writevelocity = 0 and writeall = 0.
Overall format
The saved .cn files have the following format.
- The first line is the number of atoms NP.
- This is followed by NP lines of data, each corresponding to an atom (more explanation below).
- After that, there is a 3x3 matrix
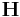 whose column vectors are the three repeat vector of the supercell.
whose column vectors are the three repeat vector of the supercell.
- The next line contains the number of species and the name of the species. By default, the number of species is 1 and the name of the species is "Mo". The species name can be set in the input file by, e.g. element0 = "Si", element1 = "C". The species names will not be used by the simulation. Even if you forgot to set them, the simulation can still run correctly.
- The last line contains two numbers: zeta and zetav. These are variables used in the Nose-Hoover thermostat. We save them to enhance reproducibility of MD simulations from a saved .cn (restart) file. These values are zero if the file is saved after quasi-static simulations.
Basic atomic information
When writeall = 0 and writevelocity = 0 (default), each line in the saved .cn file (from line 2 to line NP + 1) has the following format.
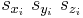
They are the scaled coordinates of atom i.
If we let 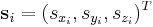 be the scaled coordinate vector of atom i and let
be the scaled coordinate vector of atom i and let 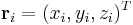 be the real coordinate vector, then they are related to each other by,
be the real coordinate vector, then they are related to each other by,
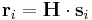
where is the matrix described in the previous sub-section.
The default setting is sufficient for quasi-static (energy minimization) simulations involving a single species without any fixed atoms.
Use writevelocity
If we want to save a restart file from Molecular Dynamics simulations, we need to save atomic velocities. This can be turned on by setting writevelocity = 1, as in the following command.
finalcnfile = myfile.cn writevelocity = 1 writecn
The command for reading this file is the same as before. MD++ will automatically detect the format of the saved .cn files.
When writevelocity = 1, each line in the saved .cn file (from line 2 to line NP + 1) has the following format.
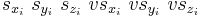
The last three numbers are the scaled velocities of atom i. They are related to the real velocities in the same way as the scaled coordinates are related to the real coordinates.
Use writeall
If the simulation contains more than one species, or if some atoms are fixed, these kinds of information can only be saved if we set writeall = 1, as in the following command.
finalcnfile = myfile.cn writeall = 1 writecn
If writeall = 1, atomic velocities will be written to file regardless of the value of the writevelocity varible.
When writeall = 1, each line in the saved .cn file (from line 2 to line NP + 1) has the following format.
![s_{x_i} \ s_{y_i} \ s_{z_i} \ vs_{x_i} \ vs_{y_i} \ vs_{z_i} \ {\rm epot[}i{\rm ]} \ {\rm fixed[}i{\rm ]} \ {\rm topol[}i{\rm ]} \ {\rm species[}i{\rm ]} \ {\rm group[}i{\rm ]} \ {\rm image[}i{\rm ]}](/mediawiki/images/math/a/d/e/adebb837da6eb3b970a75c54dc278521.png)
The last 6 numbers are additional attributes of atom i in MD++.
- epot[i] is the local potential energy of atom i. The sum epot[i] for all atoms equals to the potential energy of the system.
- fixed[i] has the default value 0. If it equals to 1, then atom i is fixed. Its position will not change in energy minimization or Molecular Dynamics simulations. If it equals to -1, then this atom is "removed". It will not be included in the evaluation of potential energy, and it will not exert any force on other atoms. (This is a useful way to remove an atom without changing the indices of other atoms.)
- topol[i] stores the central symetry deviation (CSD) parameter of atom i or other parameters computed by visualization algorithms. By default, it is not computed. To turn on the calculation of this field in energy minimzation or MD simulations, set plot_color_axis = 2. You can also ask MD++ to compute the topol[i] value for the current configuration by calling calcentralsymmetry.
- species[i] stores the element type of atom i. For a binary system, species[i] is either 0 or 1. For a ternary system, species[i] is either 0, 1 or 2. The interatomic potential function uses this field to decide which function to use, e.g. to compute the interaction between two given atoms.
- group[i] specifies which group atom i belongs to. By default, group[i] = 0 for all atoms. One way to define a group of atoms is to use commands fixatoms_by_position or fixatoms_by_ID, followed by setfixedatomsgroup and freeallatoms. Once a group (or several groups) of atoms are defined, we can use commands movegroup, setgroupcomvel, addFext_to_group, relax_by_group to manipulate them.
- image[i] has the default value -1. If it is not -1, it means atom i is not a real atom but a (periodic) image of another atom, whose index is stored in image[i]. This means that if atom image[i] moves in one step of the simulation, MD++ will move atom i by the same amount before going to the next step.
Property .out File
An MD simulation will produce a property file if saveprop = 1 is set before the run command, as is given in the following example.
saveprop = 1 savepropfreq = 100 output_fmt = "curstep EPOT KATOM Tinst TSTRESSinMPa_zz H_33" outpropfile = prop.out openpropfile run
(This is written for the .script input format. For .tcl input format, we need to add MD++ at the beginning of each line.)
In this example, one line of information is written into file prop.out every 100 MD steps. The content is specified by the output_fmt variable. Every variable that can be set in an MD++ input script can be included in output_fmt. The following is a brief explanation of the few variables included in this variable.
- curstep is the current step of the simulation.
- EPOT is the potential energy of the system.
- KATOM is the kinetic energy of all atoms. EPOT + KATOM is the total energy and should be conserved in an NVE MD simulation.
- Tinst is the instantaneous temperature in K.
- TSTRESSinMPa_zz is the zz component of the total stress given by the Virial formula, including both kinetic and potential energy terms. MD++ follows the sign convention of Parrinello-Rahman, which is opposite to many elasticity books. In MD++, a normal stress component has positive value if it is compressive.
- H_33 is the 33 component of the matrix.