Manual 05 for MD++
MD++ Tour with Sample Scripts
Keonwook Kang and Wei Cai
[First written, [ Jan 19 ]], 2007
[Last Modified, [ Feb 25 ]], 2008
Contents |
Finite Temperature Simulation
In Manual 02, we learned how to create a perfect BCC, FCC or DC crystal by the MD++ command makecrystal, by which we assigned the initial position to each of the atoms. For nonzero finite temperature simulation or molecular dynamics (MD) simulation, we need to additionally give the initial velocities and set parameters such as numerical integrator, time step and so on. In this manual, we aim to be familiar with MD++ commands and variables related with MD simulations. MD simulations can be categorized as microcanonical (NVE), canonical (NVT), isoenthalpic-isobaric (NPH) or constant pressure/temperature (NPT) ensemble type according to which physical quantities are expected to be constant or controlled. For example, a system of microcanonical ensemble has the total energy (E), the system volume (V) and the number of atoms (N) to be conserved during MD simulation. Basically, you need to choose a proper ensemble set for your own purpose of simulation.
Microcanonical(NVE) Ensemble
It would be a good start explaining MD simulation of NVE ensemble to understand what you do with MD simulations. Imagine a system of N-particle gas inside of a rigid container which is well insulated that the system is isolated from the environment. There is no heat and mass transfer through the container wall. Then you expect that the number of particles, the container volume and the total energy will not change. In MD simulations, you do not see a visible wall but you may think of a closed system with fictitious walls.
Now I give the example script mo_NVE.tcl below, written in Tcl version, which shows how to run typical NVE MD simulation from the perfect BCC molybdenum (Mo) structure. You can run the script by typing
$ bin/fs_gpp scripts/mo_NVE.tcl
# -*-shell-script -*-
# run NVE MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
proc initmd { } { MD++ {
#setnolog
setoverwrite
dirname = runs/mo-example
zipfiles = 1 # zip output files
#
#--------------------------------------------
# Create Perfect Crystal
element0 = Mo
crystalstructure = body-centered-cubic
latticeconst = 3.1472 # in Angstrom for Mo
latticesize = [ 1 0 0 5
0 1 0 5
0 0 1 5]
}}
#------------------------------------------------------------
proc readMoPot { } { MD++ {
# Read the potential file
potfile = ~/Codes/MD++/potentials/mo_pot readpot
}}
#-------------------------------------------------------------
proc openwindow { } { MD++ {
# Plot Configuration
atomradius = 1.0 bondradius = 0.3 bondlength = 2.725
atomcolor = orange highlightcolor = purple
bondcolor = red backgroundcolor = gray70
plotfreq = 10 rotateangles = [ 0 0 0 1.25 ]
openwin alloccolors rotate saverot eval plot
}}
#--------------------------------------------
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NVE" integrator_type = "VVerlet"
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (in g/mol)
timestep = 0.001 # (in ps)
totalsteps = 2000
saveprop = 1 savepropfreq = 10
savecn = 0 savecnfreq = 100
writeall = 1
DOUBLE_T = 1 randseed = 12345 srand48
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ makecrystal finalcnfile = perf.cn writecn
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ initvelocity finalcnfile = init.cn writecn
MD++ {output_fmt = "curstep EPOT KATOM Tinst"}
MD++ outpropfile = thermo.out openpropfile
MD++ run closepropfile
MD++ finalcnfile = 300K-5X5X5.cn writecn eval
MD++ sleep quit
}}
In the fuction setup_md { } in the above script, important MD++ parameters for MD simulations are assigned. First, you choose which ensemble you impose by specifying ensemble_type to be NVE, NVT, NPH or NPT. You also decide the integrator between Gear6 predictor-corrector and Verlet by saying integrator_type = "Gear6" or integrator_type = "Verlet". The predictor-corrector integrator has higher order of accuracy but the Verlet integrator is symplectic and is believed to be resilient to the larger time step.
atommass is the atomic mass in g/mol, timestep is Δt used in the integrator in the unit of picosecond, totalsteps is the total number of iterations in time integration. As long as it is guaranteed that the total energy of the system is conserved, the timestep (timestep) is usually taken as big as possible. The total time step (totalsteps) is typically chosen long enough for the system to reach its dynamical steady state.
T_OBJ is the target temperature in Kelvin in NVT ensemble (which will be explained in the next section), but it is used here just to generate atoms' initial velocities and that does not necessarily mean the temperature will be maintained at the specified number during the simulation because there is no temperature we control in NVE ensemble. The MD++ command initvelocity initializes the velocity to each atom randomly according to the value of T_OBJ. For random number generation, srand48 is executed with a random seed randseed.
If the parameter DOUBLE_T is set to be 1, the initial temperature becomes twice. You can see the effect of DOUBLE_T = 1 in Fig.1. When you see the temperature plot in Fig.1, you will realize that the temperature at equilibrated state seems to be half of the initial temperature. The reason why the temperature reduces by half is followed. Since the initial atomic configuration at 0 K is the local-energy-minimum structure, part of the kinetic energy initially given is always converted to the potential energy during finite temperature simulation and this portion amounts to be half of the kinetic energy.
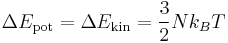 . .
|
for 3D case due to the equipartition theorem.
- Fig.1 The effect of DOUBLE_T
Temperature in NVE ensemble with different DOUBLE_T setting

The MD++ command run starts MD simulation with all these parameters. There are more parameters related with MD simulation. saveprop and savepropfreq will determine whether or not the thermodynamic properties will be written in file and how often they will be. If saveprop = 1 and savepropfreq = 10, the thermodynamic properties such as potential energy, kinetic energy and temperature will be dumped out at every 10 step only if the MD++ command openpropfile is executed.
The format of a property file is defined by output_fmt. Accordig to the example script, each line of the property file contains current step (curstep), potential energy (EPOT), kinetic energy (KATOM), and temperature (Tinst). If we do not specify output_fmt, the default array of thermodynamic data output is current step (curstep), kinetic energy (KATOM), potential energy (EPOT), pressure (PRESSURE), -σxy (TSTRESS_xy), -σyz (TSTRESS_yz), -σzx (TSTRESS_zx), HELM, the extended energy(HELMP), thermodynamic friction coefficient (zeta), dEdlambda and the system volume (OMEGA). Units are given as eV for energy and eV/A˚^3 for stress. The sign of the stress TSTRESS is the opposite of the conventional definition in elasticity or continuum theory, because MD++ is implemented following Virial's definition of stress.
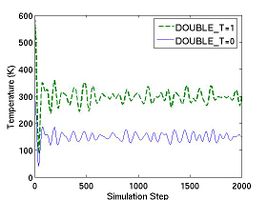
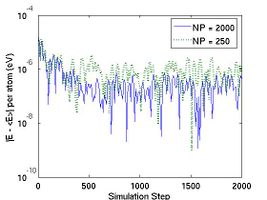
The thermodynamic properties can be loaded and plotted by the external program such as Octave and Matlab. In Fig.2, total energy per atom and temperature are plotted for the systems of two different size. The bigger system (10[100] X 10[010] X 10[001]) has eight times more atoms than the other (5[100] X 5[010] X 5[001]). Obviously, you see that the thermodynamic data in the bigger system show less fluctuation.
- Fig.2 Energy and Temperature fluctuation of NVE ensemble
(a) Energy Fluctuation in NVE ensemble.
(b) Temperature in NVE ensemble


savecn and savecnfreq are used to determine how often the intermediate configuration will be saved as .cn file with the MD++ command openintercnfile just like saveprop and savepropfreq. You can specify names of the property file and the intermediate configuration files using MD++ parameter outpropfile and intercnfile, respectively. If we do not specify them, the default file name would be prop.out and inter.cn. The expression zipfiles = 1 in the 11th line will compress files such as .out and .cn. For example, the configuration file init.cn and the thermodynamic data file thermo.out will be zipped as init.cn.gz and thermo.out.gz, respectively.
A configuration (.cn) file contains the following informations: the number of atoms (NP) in the first line and a 3 by 3 periodicity matrix 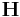 in the last three lines. Inbetween, the scaled cooridinates (_SR[i].x, _SR[i].y and _SR[i].z for i=1 to NP) of NP atoms are stored in default. If writeall = 1, additional informations are added such that each line has sacaled velocities (_VSR[i].x, _VSR[i].y and _VSR[i].z), individual potential energy (_EPOT_IND[i]), whether-or-not fixed (fixed[i]), topology (_TOPOL[i]), atom species (species[i]), atom groups (group[i]) and images (image[i]) after the scaled coordinates. You can check this difference by comparing two configuration files, perf.cn and init.cn. Because perf.cn is written before setting writeall = 1, it has only number of atoms, scaled coordinates of each atom and the box matrix while init.cn has all the informations. Alternatively, writevelocity = 1 can be used, which stores only the scaled coordinates and velocities.
in the last three lines. Inbetween, the scaled cooridinates (_SR[i].x, _SR[i].y and _SR[i].z for i=1 to NP) of NP atoms are stored in default. If writeall = 1, additional informations are added such that each line has sacaled velocities (_VSR[i].x, _VSR[i].y and _VSR[i].z), individual potential energy (_EPOT_IND[i]), whether-or-not fixed (fixed[i]), topology (_TOPOL[i]), atom species (species[i]), atom groups (group[i]) and images (image[i]) after the scaled coordinates. You can check this difference by comparing two configuration files, perf.cn and init.cn. Because perf.cn is written before setting writeall = 1, it has only number of atoms, scaled coordinates of each atom and the box matrix while init.cn has all the informations. Alternatively, writevelocity = 1 can be used, which stores only the scaled coordinates and velocities.
Commands and variables related with visualization in openwindow will be covered in a separate manual in detail.
Canonical(NVT) Ensemble
In the following example script mo_NVT.tcl, BCC molybdenum (Mo) will be simulated at 300 K using Nose-Hoover thermostat<ref> S. Nose, “A Molecular Dynamics Method for Simulations in the Canonical Ensemble”, Molecular Physics, 52 255 (1984)</ref>. Run the script by typing
$ bin/fs_gpp scripts/mo_NVT.tcl
# -*-shell-script -*-
# run NVT MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
proc initmd { } {
...
}
#------------------------------------------------------------
proc readMoPot { } {
...
}
#-------------------------------------------------------------
proc openwindow { } {
...
}
#--------------------------------------------
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NVT" integrator_type = "VVerlet"
implementation_type = 0 vt2 = 1e28
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (g/mol)
timestep = 0.001 # (ps)
totalsteps = 2000
saveprop = 1 savepropfreq = 10
savecn = 0 savecnfreq = 100
writeall = 1
}}
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ {
incnfile = 300K-5X5X5.cn
atommass = 95.94 timestep = 0.001
readcn
}
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ {output_fmt = "curstep EPOT KATOM Tinst HELMP"}
MD++ outpropfile = thermo-NVT.out openpropfile
MD++ run closepropfile
MD++ finalcnfile = 300K-5X5X5-NVT.cn writecn eval
MD++ sleep quit
Instead of creating BCC Mo crystal, this script reads the configuration file 300K_5X5X5.cn, which was generated by the previous script mo_NVE.tcl, using the MD++ command readcn. Note that when you read the configuration file, the timestep should be same with that of the previous run because the scaled velocity is stored as dimensionless unit with the timestep multiplied. It is safe that you specify timestep and atommass when reading .cn file as shown in the script.
Setting ensemble_type = "NVT" activates Nose-Hoover thermostat. In MD++, there are three different implementations of NVT integrator defined through implementation_type. If it is zero, the implicit integrator will be chosen. If it is one, the explicit integrator based on Sto¨rmer-Verlet method will work. If it is two, another explicit integrator based on Liouville formulation will be activated.
Thermal coefficient vt2 is related with thermal mass Q of Nose´-Hoover thermo-stat as
Isoenthalpic-isobaric (NPH) Ensemble