Manual 04 for MD++
Creating a Vacancy, Relaxing The Imperfect Structure
and Calculating Vacancy Formation Energy
Keonwook Kang and Wei Cai
Contents |
Creating a Vacancy in a Perfect Crystal
A vacancy is a common and important type of point defect in solids. It is an empty site (i.e. a missing atom) in an otherwise prefect crystal structure. Here we discuss how to introduce a vacancy in a perfect crystal using MD++ and how to compute the vacancy formation energy. Consider the following MD++ input file, movacancy.script, that creates a vacancy in a perfect BCC Molybdenum crystal.
# -*-shell-script-*-
setnolog
setoverwrite
dirname = runs/movacancy # specify run directory
#--------------------------------------------
# Read in potential file
potfile = ~/Codes/MD++/potentials/mo_pot readpot
#--------------------------------------------
#Create Perfect Lattice Configuration
crystalstructure = body-centered-cubic latticeconst = 3.1472 #(A)
latticesize = [ 1 0 0 5
0 1 0 5
0 0 1 5]
makecrystal finalcnfile = perf.cn writecn
eval # evaluate the potential of perfect crystal
#--------------------------------------------
# Create Vacancy
input = [ 1 # number of atoms to be fixed
0] # index of an atom to be fixed
fixatoms_by_ID # fix a set of atoms by their indices
removefixedatoms # remove fixed atoms
finalcnfile = movac.cn writecn
eval # evaluate the vacancy-formed crystal
#---------------------------------------------
# Plot Configuration
atomradius = 1.0 bondradius = 0.3 bondlength = 0
atomcolor = blue highlightcolor = purple backgroundcolor = gray
bondcolor = red fixatomcolor = yellow
plotfreq = 10 win_width = 600 win_height = 600
plot_atom_info = 3
color00 = "orange" color01 = "purple" color02 = "green"
color03 = "magenta" color04 = "cyan" color05 = "purple"
color06 = "gray80" color07 = "white"
plot_color_windows = [ 2 # number of color windows
-10 -6.8 6 # color06 = gray80
-6.7 -6.0 0 # color00 = orange
]
rotateangles = [ 0 0 0 1 ]
openwin alloccolors rotate saverot plot
sleep quit
We can run this script file by typing
$ bin/fs gpp scripts/movacancy.script
First, MD++ creates a 5×5×5 perfect cubic crystal of Mo with edges along <100> directions. Then it fixes atom 0 by
input = [ 1 # number of atoms to be fixed
0 ] # index of an atom to be fixed
fixatoms_by_ID
and then removs this atom by the command removefixedatoms. The first number in the input array specifies the number of atoms to be removed. For example, if you would like to remove two atoms, say 3 and 8, you can do so by
input = [ 2 # number of atoms to be fixed
3 8 ] # index of atoms to be fixed
fixatoms_by_ID # fix a set of atoms by their index
removefixedatoms # remove fixed atoms
Sometimes, you may want to remove a specific atom you see in the graphic window. You can obtain the index of this atom by clicking the atom with your mouse. Depending on the setting of plot_atom_info, other information is also displayed when you click on the atom. If plot_atom_info = 1, the atom index number and its scaled coordinates are printed when the atom is clicked. If plot_atom_info = 2, the atom index and its real coordinate (in Å) will be printed. If plot_atom_info = 3, the atom index and its local energy (in eV) will be printed.
The script file also sets up two color windows (plot_color_windows) to display the atoms. Atoms with local energy between -10 eV and -6.8 eV are shown in gray and the atoms whose energies lie between -6.7 eV and -6.0 eV are shown in orange. This will highlight the atoms near the vacancy because they usually have a higher local energy. Click the atoms and you will see the actual local energy of these atoms. If you compare the potential energy of the perfect structure and the vacancy-formed structure, you will know which structure has higher energy.
Q.1 If a single atom, whose index is other than 0, is removed from the same perfect structure, do we expect the system to have a different potential energy?
Relaxation
After creating a vacancy, it is not hard to imagine that the atoms around the vacancy will not stay as they were, for they have less bonds and form a locally unstable structure. We can expect the atoms near the vacancy will move toward the vacant site to some degree and lower the total energy of the system. In other words, the atomistic structure will be relaxed to its locay energy minimum state from the given initial position. MD++ uses a conjugate gradient relaxation (CGR) algorithm<ref> Numerical Recipes, http://www.nr.com/</ref> for this energy minimization process. for which the script file needs the section given below.
#--------------------------------------------- # Conjugate-Gradient relaxation conj_ftol = 1e-7 # tolerance on the residual gradient conj_fevalmax = 1000 # max. number of iterations conj_fixbox = 1 # fix the simulation box relax # CG relaxation command finalcnfile = relaxed.cn writecn eval # evaluate relaxed structure
Let’s copy the above script and paste it to the movacancy.script right before the last command, sleep quit. You will watch the potential energy of the simulated system keeps decreasing while the relaxation goes on. There are a coulple of variables related with CG relaxation. conj_ftol is the tolerance of the residual gradient and conj_fevalmax is the maximum number of iterations. The relaxtion will stop if the tolerance becomes smaller than the specified conj_ftol or if the number of iteration reaches its maximum. If conj_fixbox is equal to 1, the shape and size of simulation cell box is fixed so that only the atoms can move during the relaxation. Sometimes you need to relax the simulation box together with atoms. In such a case you can add more degrees of freedom to the system by setting conj_fixbox = 0. For example, you can allow the box to expand or shrink hydrostatically by saying that
conj_fixbox = 0
conj_fixboxvec = [ 0 1 1
1 0 1
1 1 0]
In this case, only the diagonal components of the box matrix 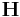 can relax and
the other components are fixed as assigned to conj_fixboxvec.
can relax and
the other components are fixed as assigned to conj_fixboxvec.
Vacancy Formation Energy
We define the vacancy formation energy Ev as the energy needed to create a vacancy from the bulk perfect crystal structure. This can be obtained by comparing the energy of the relaxed vacancy-formed structure and the energy of a perfect crystal structure. However, it is not just a simple substraction between two, because we need to make sure we compare two systems of the same number of atoms. In reality, no atom is removed or destroyed when a vacancy forms. Rather, what we can think of is displacement of an inside atom to the free surface of a solid leaving a vacancy inside as shown in Fig. 2(a). Even though there is no free surface in our simulation with the periodic boundary condition. Let N be the total number of atoms in the original perfect crystal structure. And E1 is the energy of the perfect crystal and E2 is the energy of relaxed structure of (N1) atoms. Then the vacancy formation energy Ev can be expressed as
N −1
Ev ≡ E2 − · E1 .
N
Q.2 Can you tell the different physical meanings between two expres
sions 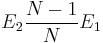 and E2E1? You can use Fig. 2 for your explanation.
and E2E1? You can use Fig. 2 for your explanation.
Once you know the vacancy formation energy Ev, you can use it to estimate the concentration of vacancy <maht>[n]</math> in the crystal strucutre at given temperature T and atmospheric pressure from Boltzmans distribution relation<ref> R. J. Stokes and D. F. Evans, Fundamentals of Interfacial Engineering, Wiley-VCH 1997 pp.491-493 </ref>,
[n] ∼ exp(−Ev /kB T )
where kB = 8.621E-5 eV/K is Boltzman’s constant. For example, the vacancy formation energy of Mo is 2.550 (eV)<ref> With the tight binding model, it is reported to be 2.46 eV. See the referene, M. J. Mehl and D. A. Papaconstantopoulos, “Applications of a tight-binding total-energy method for transition and noble metals: Elastic constants, vacancies, and surfaces of monatomic metals”, Physical Review B 54 4519-4530 1996 </ref> from the atomistic calculation. Then the fraction of vacancy will be on the order of 1.514E-43 at 300 K.
Q.3 Calculate the vacancy formation energy of Mo with the simulation box of different size. The vacancy formation energy is expected to be the same irrelavant to the box size. However, you will find the energy dramatically changes if the box is smaller than the certain size. Explain the reason.