M05 Finite Temperature Simulation: Difference between revisions
m (introduction) |
|||
| (16 intermediate revisions by 2 users not shown) | |||
| Line 7: | Line 7: | ||
</DIV> |
</DIV> |
||
<P ALIGN="CENTER">[First written, [ Jan 19 ]], [[2007]]</P> |
<P ALIGN="CENTER">[First written, [ Jan 19 ]], [[2007]]</P> |
||
<P ALIGN="CENTER">[Last Modified, [ |
<P ALIGN="CENTER">[Last Modified, [ Apr 13 ]], [[2008]]</P> |
||
<P> |
<P> |
||
<BR><HR> |
<BR><HR> |
||
<!--Table of Child-Links--> |
<!--Table of Child-Links--> |
||
The case studies described in Manuals 02-04 only deal with atomic positions as degrees of freedom. For example, in Manual 02, we learned how to create a perfect crystal by MD++ command '''makecrystal'''. In Manual 03, we varied the size of the perfect crystal to find the equilibrium lattice constants. In Manual 04, we introduced a vacancy to the perfect crystal and let the atomic positions relax to a local energy minimum. In this manual, we will learn how to perform finite temperature, Molecular Dynamics (MD) simulations. For this purpose, we will need to deal with atomic velocities as degrees of freedom, as well as a number of control variables, such as integrator type, time step, and output controls. We will learn to specify the simulation ensembles from |
The case studies described in Manuals 02-04 only deal with atomic positions as degrees of freedom. For example, in Manual 02, we learned how to create a perfect crystal by MD++ command '''makecrystal'''. In Manual 03, we varied the size of the perfect crystal to find the equilibrium lattice constants. In Manual 04, we introduced a vacancy to the perfect crystal and let the atomic positions relax to a local energy minimum. In this manual, we will learn how to perform finite temperature, Molecular Dynamics (MD) simulations. For this purpose, we will need to deal with atomic velocities as degrees of freedom, as well as a number of control variables, such as integrator type, time step, and output controls. We will learn to specify the simulation ensembles from different choices: microcanonical (NVE), canonical (NVT), |
||
isoenthalpic-isobaric (NPH) and constant pressure/temperature (NPT) ensembles. Different quantities are conserved or controlled in different ensembles. For example, the total number of atoms (N), volume (V) and energy (E) are conserved in the (NVE) ensemble. |
isoenthalpic-isobaric (NPH) and constant pressure/temperature (NPT) ensembles. Different quantities are conserved or controlled in different ensembles. For example, the total number of atoms (N), volume (V) and energy (E) are conserved in the (NVE) ensemble. |
||
=== Microcanonical (NVE) Ensemble === |
=== Microcanonical (NVE) Ensemble === |
||
<HR> |
<HR> |
||
The NVE ensemble is the probably the best starting point to understand what happens in MD simulations. In Statistical Mechanics, to picture the NVE ensemble, we can imagine a system of N gas molecules inside a rigid container with perfect thermal insulation. Hence no work or heat can be exchanged between the system and the outside world (its environment). While the gas molecules move with time, the total number of particles N, the total energy E and the total volume V of the system does not change. A large number of replicas of the system with the same N, V, E forms the NVE, or microcanonical ensemble in Statistical Mechanics. In MD simulations within the NVE ensemble, we usually do not simulate the rigid container --- this is because the simulation volume is usually very small and having an explicit container will lead to a large surface-to-volume ratio and large simulation artifacts. Instead, periodic boundary conditions (PBC) are usually used in MD simulation. If the repeat vectors of PBC do not change, the simulation volume V is kept constant during the simulation. Since the particles follows Hamiltonian dynamics in MD simulations, the total energy E is conserved. The total number of particles N is obviously conserved in the MD simulation. Therefore, MD simulations under PBC with fixed repeat vectors and no thermostats (to be described later) correspond to the NVE (microcanonical) ensemble. |
|||
It would be a good start explaining MD simulation of NVE ensemble to understand what you do with MD simulations. |
|||
Imagine a system of N-particle gas inside of a rigid container which is well insulated that the system is isolated from the environment. |
|||
There is no heat and mass transfer through the container wall. Then you expect that the number of particles, the container volume and the total energy will not change. In MD simulations of NVE ensemble, you do not see a visible wall but you may think of a closed system with fictitious walls. |
|||
The following input script <tt>mo_NVE.tcl</tt> gives an example of running MD simulations in MD++. You can test is by the following command (if you put this file in your <tt>scripts/</tt> directory) |
|||
Now I give the example script ''mo_NVE.tcl'' below, written in Tcl version. You can run the script by typing |
|||
$ bin/fs_gpp scripts/mo_NVE.tcl |
$ bin/fs_gpp scripts/mo_NVE.tcl |
||
This example script first creates a perfect BCC crystal of molybdenum (Mo) with a supercell of 5[1 0 0] <math>\times</math> 5[0 1 0] <math>\times</math> 5[0 0 1]. It then runs MD simulations using the Finnis-Sinclair (FS) potential. |
|||
<pre> |
<pre> |
||
# -*-shell-script -*- |
# -*-shell-script -*- |
||
| Line 75: | Line 74: | ||
writeall = 1 DOUBLE_T = 1 |
writeall = 1 DOUBLE_T = 1 |
||
randseed = 12345 srand48 #randomize random number generator |
randseed = 12345 srand48 #randomize random number generator |
||
}} |
|||
#******************************************* |
#******************************************* |
||
| Line 93: | Line 93: | ||
MD++ finalcnfile = 300K-5X5X5.cn writecn eval |
MD++ finalcnfile = 300K-5X5X5.cn writecn eval |
||
MD++ sleep quit |
MD++ sleep quit |
||
}} |
|||
</pre> |
</pre> |
||
In the fuction '''setup_md { }''' in the above script, important MD++ parameters for MD simulations are assigned. First, you choose which ensemble you impose by specifying '''ensemble_type''' to be '''NVE''', '''NVT''', '''NPH''' or '''NPT'''. You also decide the integrator between Gear 6-th order predictor-corrector and velocity Verlet by saying '''integrator_type = "Gear6"''' or '''integrator_type = "VVerlet"'''. The predictor-corrector integrator has higher order of accuracy but the Verlet integrator is symplectic and is believed to be resilient to the larger time step. |
|||
In this Tcl script, we start with some definition of functions that will be used subsequently. The main program starts after the comment <tt>Main program starts here</tt>. The program first calls the <tt>initmd</tt> function that was defined at the beginning, which opens the output directory. After creating the perfect crystal using the <tt>makecrystal</tt> command, it calls the <tt>setup_md</tt> function that initializes some important parameters for MD simulation. (Commands and variables related with visualization as specified in the <tt>openwindow</tt> function will be covered in a separate manual in detail.) |
|||
'''atommass''' is the atomic mass in g/mol, '''timestep''' is <math>\Delta t</math> used in the integrator in the unit of picosecond, '''totalsteps''' is the total number of iterations in time integration. As long as it is guaranteed that the total energy of the system is conserved, the timestep ('''timestep''') is usually taken as big as possible. The total time step ('''totalsteps''') is typically chosen long enough for the system to reach its dynamical steady state. |
|||
The initial velocity is first assigned as random numbers but then scaled to correspond to an instantaneous temperature of '''T_OBJ'''.<ref>'''T_OBJ''' is the target temperature in NVT ensemble. However, it is used here just to generate atoms' initial velocities and that does not necessarily mean the temperature will be maintained at the specified value during the simulation because there is no temperature we control in NVE ensemble.</ref> The MD++ command '''initvelocity''' initializes the velocity to each atom randomly according to the value of '''T_OBJ'''. For random number generation, '''srand48''' is executed with an integer random seed '''randseed'''. Running the simulation again with the same '''randseed''' will give you the same result (which is good for debugging). |
|||
Running the simulation again with a different '''randseed''' will give you a different initial velocity condition (which is good for collecting statistics). You may also use the MD++ command '''srand48bytime''', which will use the current time as the random seed (in this way, your simulation will always have a different initial velocity condition whenever you run it again). |
|||
The first line in the <tt>setup_md</tt> function selects the statistica ensemble for the simulation. The possible choices for the variable '''ensemble_type''' are '''NVE''', '''NVT''', '''NPH''' and '''NPT'''. The type of the numerical integrator is specified by the '''integrator_type''' variable and can be either the Gear 6-th order predictor-corrector algorithm ('''Gear6''') or the velocity Verlet algorithm ('''VVerlet'''). The predictor-corrector integrator has a higher order of accuracy but velocity Verlet is a symplectic integrator and is more stable at larger algorithm. We recommend the use of velocity Verlet algorithm whenever possible (i.e. if it is implemented for the chosen ensemble type). |
|||
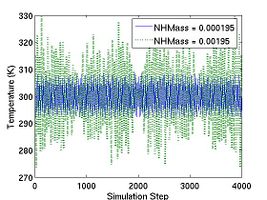
If the parameter '''DOUBLE_T''' is set to be 1, the initial temperature becomes twice. You can see the effect of '''DOUBLE_T = 1''' in Fig.1. When you see the temperature plot in Fig.1, you will realize that the temperature at equilibrated state seems to be half of the initial temperature. The reason why the temperature reduces by half is followed. Since the initial atomic configuration at 0 K is the local-energy-minimum structure, part of the kinetic energy initially given is always converted to the potential energy during MD simulation and this portion amounts to be half of the kinetic energy. |
|||
{|border="0" align="center" |
|||
The variable '''atommass''' specifies the atomic mass in unit of g/mol. '''timestep''' specifies the integrator time step <math>\Delta t</math> in unit of picosecond. '''totalsteps''' is the total number of time steps for the MD simulation. Hence '''totalsteps''' times '''timestep''' is the total time duration of the MD simulation. We usually choose '''timestep''' to be as large as possible provided that the total energy of the system is conserved within an acceptable accurady. '''totalsteps''' is usually chosen so that the total simulation time is long enough for the system to reach an equilibrium or steady state (usually on the order of picoseconds). |
|||
|<math> \Delta E_{\mathrm{pot}} = \Delta E_{\mathrm{kin}} = \frac{3}{2} N k_B T</math>. |
|||
|} |
|||
The command <tt>initvelocity</tt> assign random numbers to the atomic velocities and then scale them so that the instantaneous temperature matches the target value, as specified by '''T_OBJ''' in unit of K. Strictly speaking, temperature is not well defined in an NVE ensemble (but is well defined in the NVT ensemble). Here we can regard '''T_OBJ''' as a measure of the instantaneous kinetic energy of the system. MD++ uses the <tt>drand48()</tt> to generate random numbers for the velocities. The random number generator can be initialized by calling <tt>srand48</tt> with a specified '''randseed'''. This approach is more convenient for debugging because the same '''randseed''' garauntees exactly the same random number sequence will be generated if you run the simulation again. On the other hand, you may use the function <tt>srandbytime</tt> to use the current time as the random seed for initializing the random number generator. This makes sure that ever time you run the simulation again a completely different set of random numbers will be generated. |
|||
for 3D case due to the equipartition theorem. |
|||
If the parameter '''DOUBLE_T''' is set to 1, then the velocities are scaled in such a way that the instantaneous temperature is twice of '''T_OBJ''', as illustrated in Fig.1. The reason we may want to set <tt>DOUBLE_T = 1</tt> is the following. When we assign velocities to the atoms in a perfect crystal, the instantaneous temperature almost always drop to half of its initial value when thermal equilibrium is reached (see Fig.1). This is due to the energy equipartition theorem. For solids at temperatures much below the melting temperature, the Hamiltonian is close to that of a set of coupled harmonic oscillators. The total energy of a harmonic system is equally divided between the average kinetic energy and the average potential energy. Because the total energy is conserved in an NVE-ensemble simulation, the kinetic energy is bound to decrease by half if the kinetic energy and potential energy (initially zero) are equal (in time average) when equilibrium is reached. Setting <tt>DOUBLE_T = 1</tt> allows the temperature at the equilibrium state to match the desired temperature. |
|||
<gallery caption="Fig.1 |
<gallery caption="Fig.1 Instantaneous temperature as a function of time during MD simulations with different settings for DOUBLE_T." widths="300px" heights="200px" perrow="1"> |
||
Image:Temp_double.jpg |
Image:Temp_double.jpg |
||
</gallery> |
</gallery> |
||
The MD++ command '''run''' starts MD simulation with all these parameters. There are more parameters related with MD simulation. '''saveprop''' and '''savepropfreq''' will determine whether or not the thermodynamic properties will be written in file and how often they will be. If '''saveprop = 1''' and '''savepropfreq = 10''', the thermodynamic properties such as potential energy, kinetic energy and temperature will be dumped out at every 10 step only if the MD++ command '''openpropfile''' is executed. |
|||
The parameters '''saveprop''' and '''savepropfreq''' specify whether or not and how often the simulation properties will be saved periodically in a data file. If <tt>saveprop = 1</tt> and <tt>savepropfreq = 10</tt>, the properties such as potential energy, kinetic energy and temperature will be save every 10 simulation steps, provided that the MD++ command '''openpropfile''' is called before the command '''run'''. The name of the property file is specified by the '''outputfile''' variable and is <tt>prop.out</tt> by default. The setting <tt>zipfiles = 1</tt> at the beginning of this script file (line 11) specifies that both property files and atomic configuration files (see below) will be automatically zipped (by gzip) after they are written. |
|||
The content of a property file is defined by '''output_fmt'''. Accordig to the example script, each line of the property file contains current step ('''curstep'''), potential energy ('''EPOT'''), kinetic energy ('''KATOM'''), and temperature ('''Tinst'''). If we do not specify '''output_fmt''', the default array of thermodynamic data output is current step ('''curstep'''), kinetic energy ('''KATOM'''), potential energy ('''EPOT'''), pressure ('''PRESSURE'''), -<math>\sigma_{xy}</math> ('''TSTRESS_xy'''), -<math>\sigma_{yz}</math> ('''TSTRESS_yz'''), -<math>\sigma_{zx}</math> ('''TSTRESS_zx'''), '''HELM''', the extended energy('''HELMP'''), thermodynamic friction coefficient ('''zeta'''), '''dEdlambda''' and the system volume ('''OMEGA'''). Units are given as eV for energy and eV/A˚^3 for stress. <font color='blue'>The sign of the stress '''TSTRESS''' is the opposite of the conventional definition in elasticity or continuum theory, because MD++ is implemented following Parrinello and Rahman's definition of stress.</font><ref>M. Parrinello and A. Rahman, "Polymorphic transitions in single crystals: A new molecular dynamics method", J. Appl. Phys '''52''' 7182-7190 1981</ref> |
|||
The content of a property file is defined by variable '''output_fmt'''. In this example, each line of the property file will contain the current step ('''curstep'''), potential energy ('''EPOT'''), kinetic energy ('''KATOM'''), and temperature ('''Tinst'''). If we do not specify '''output_fmt''', the default content of the property file will be current step ('''curstep'''), kinetic energy ('''KATOM'''), potential energy ('''EPOT'''), pressure ('''PRESSURE'''), <math>\sigma_{xy}</math> ('''TSTRESS_xy'''), <math>\sigma_{yz}</math> ('''TSTRESS_yz'''), <math>\sigma_{zx}</math> ('''TSTRESS_zx'''), '''HELM''', the extended energy('''HELMP'''), thermodynamic friction coefficient ('''zeta'''), reversible work increment '''dEdlambda''' and volume ('''OMEGA'''). The energies are in unit of eV and stresses in eV/A˚^3. Please note that there is an <font color='blue'>overall minus sign</font> between the stress variables in MD++ and that in conventional elasticity theory. For example, a positive pressure correspond to positive normal stress components in MD++ but negative normal stress components in elasticity theory. This is because in MD++, we followed the sign convention of Parrinello and Rahman for stress control.<ref>M. Parrinello and A. Rahman, "Polymorphic transitions in single crystals: A new molecular dynamics method", J. Appl. Phys '''52''' 7182-7190 1981</ref> |
|||
The parameters '''savecn''' and '''savecnfreq''' specify whether or not and how often the atomic coordinates (and velocities if '''writeall = 1''') will be saved as .cn files during the MD simulation. If <tt>savecn = 1</tt> and <tt>savepropfreq = 10000</tt>, the intermediate .cn files will be saved every 10000 steps, provided that the MD++ command '''openintercnfile''' is called before the command '''run'''. The name of the intermediate .cn files are specified by the '''intercnfile''' variable and is <tt>inter####.cn</tt> by default, where #### are integers (starting with 0000) that will be automatically incremented by one after each file is written. In this example, <tt>savecn = 0</tt> so no intermediate .cn files will be saved. After all the relevant parameters have been set up, the MD++ command '''run''' starts the MD simulation. |
|||
After the simulation is finished, a property file named <tt>thermo.out</tt> will be written in the output directory <tt>runs/mo-example</tt>. This file can be loaded and plotted by programs such as Matlab, Octave, or Gnuplot.<ref>A simple way to plot the total energy using gnuplot (a free software on Unix/Linux) is to run the command. |
|||
plot "thermo.out" u ($1):($2+$3) with line |
|||
</ref> |
|||
Fig.2 plots the total energy per atom and instantanous temperature for two simulations with different sizes. The larger system (10[100] <math>\times</math> 10[010] <math>\times</math> 10[001]) has eight times more atoms than the smaller one (5[100] <math>\times</math> 5[010] <math>\times</math> 5[001]). As expected, the data for the larger system experience less statistical fluctuation. |
|||
The thermodynamic properties can be loaded and plotted by the external program such as Octave and Matlab.<ref>A simple way to plot the total energy using gnuplot (a free software on Unix/Linux) is to run the command. |
|||
plot "prop.out" u ($1):($2+$3) with line |
|||
</ref> In Fig.2, total energy per atom and temperature are plotted for the systems of two different size. The bigger system (10[100] X 10[010] X 10[001]) has eight times more atoms than the other (5[100] X 5[010] X 5[001]). Obviously, you see that the thermodynamic data in the bigger system show less fluctuation. |
|||
<gallery caption="Fig.2 Energy and Temperature fluctuation of NVE ensemble" widths="300px" heights="200px" perrow="2"> |
<gallery caption="Fig.2 Energy and Temperature fluctuation of NVE ensemble" widths="300px" heights="200px" perrow="2"> |
||
Image:Energy_fluct.jpg|(a) Energy Fluctuation in NVE ensemble. |
Image:Energy_fluct.jpg|(a) Energy Fluctuation in NVE ensemble. |
||
| Line 125: | Line 129: | ||
</gallery> |
</gallery> |
||
'''savecn''' and '''savecnfreq''' are used to determine how often the intermediate configuration will be saved as .cn file with the MD++ command '''openintercnfile''' just like '''saveprop''' and '''savepropfreq'''. You can specify names of the property file and the intermediate configuration files using MD++ parameter '''outpropfile''' and '''intercnfile''', respectively. If we do not specify them, the default file name would be '''prop.out''' and '''inter.cn'''. The expression '''zipfiles = 1''' in the 11th line will compress files such as .out and .cn. For example, the configuration file '''init.cn''' and the thermodynamic data file '''thermo.out''' will be zipped as '''init.cn.gz''' and '''thermo.out.gz''', respectively. |
|||
After the '''run''' command, the script file calls the command '''writecn''' to save the final atomic configuration into a .cn file whose name is specified by '''finalcnfile'''. The .cn file has the following format. The first line contains the total number of atoms <tt>NP</tt>. The following <tt>NP</tt> lines then contain the scaled coordinates of all atoms: <math>s_x^{(i)}</math>, <math>s_y^{(i)}</math>, <math>s_z^{(i)}</math>, <math>i = 1, 2, \cdots NP</math>. The last three lines of the .cn file specify a 3 <math>\times</math> 3 matrix <math>\mathbf{H}</math> whose column vectors are the three repeat vectors of the simulation supercell (subjected to periodic boundary conditions). The real coordinates of an atom (x, y, z ) and the scaled coordinates for each atom are related through <math>\mathbf{H}</math> as follows |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 133: | Line 136: | ||
|} |
|} |
||
If |
If <tt>writeall = 1</tt> is specified before '''writecn''', additional informations are added to the ''NP'' lines of data. Following the scaled coordinates, each line will also contain the scaled velocities ('''_VSR[i].x''', '''_VSR[i].y''' and '''_VSR[i].z'''), local potential energy ('''_EPOT_IND[i]'''), flag ('''fixed[i]'''), central-symmetry-deviation paramter ('''_TOPOL[i]'''), atom species ('''species[i]'''), atom groups ('''group[i]''') and image index ('''image[i]'''). You can see this difference by comparing two configuration files, '''perf.cn''' (written before setting <tt>writeall = 1</tt>) and '''init.cn''' (written after). If <tt>writevelocity = 1</tt> is used instead of <tt>writeall = 1</tt>, then only the scaled velocities are added behind the three columns for scaled coordinates. |
||
At the end of the example script, the command '''sleep''' is called so that the graphics window to stay open (i.e. alive) for a while so that we can examine the atomic structure. You can press ctrl-c in the terminal to exit. You can also comment out the '''sleep''' command in the script file so that MD++ exits immediately after reaching the end of the input file. |
|||
Commands and variables related with visualization in '''openwindow''' will be covered in a separate manual in detail. |
|||
After the simulation (run) has finished, the '''sleep''' command allows the graphics window to stay alive for a while so that we can look at the final atomic structure. You can press ctrl-c in the terminal to exit. You can also comment out the '''sleep''' command so that MD++ quits right after finishing '''run'''. |
|||
=== Canonical(NVT) Ensemble === |
=== Canonical(NVT) Ensemble === |
||
---- |
---- |
||
In the NVT ensemble of Statistical Mechanics, the system exchanges heat with an external thermostat to maintain its temperature at T but its total energy is no longer conserved. The following script <tt>mo_NVT.tcl</tt> provides an example of NVT simulations in MD++. You can test it by typing |
|||
The following example script '''mo_NVT.tcl''' reads the structure file of BCC Mo and runs NVT MD simulation during which temperature is maintained at 300 K using Nose-Hoover thermostat<ref> S. Nose, “A Molecular Dynamics Method for Simulations in the Canonical Ensemble”, Molecular Physics, '''52''' 255 (1984)</ref>. Run the script by typing |
|||
$ bin/fs_gpp scripts/mo_NVT.tcl |
$ bin/fs_gpp scripts/mo_NVT.tcl |
||
This script first reads in an atomic configuration generated by the previous MD simulation (NVE ensemble) and then performs MD simulation in the NVT ensemble at T = 300 K using the Nose-Hoover thermostat<ref> S. Nose, “A Molecular Dynamics Method for Simulations in the Canonical Ensemble”, Molecular Physics, '''52''' 255 (1984)</ref>. |
|||
<pre> |
<pre> |
||
# -*-shell-script -*- |
# -*-shell-script -*- |
||
| Line 150: | Line 153: | ||
# Definition of procedures |
# Definition of procedures |
||
#******************************************* |
#******************************************* |
||
# |
|||
proc initmd { } { |
|||
# ... omitted here to save space ... |
|||
# |
|||
} |
|||
#------------------------------------------------------------ |
|||
proc readMoPot { } { |
|||
... |
|||
} |
|||
#------------------------------------------------------------- |
|||
proc openwindow { } { |
|||
... |
|||
} |
|||
#-------------------------------------------- |
|||
proc setup_md { } { MD++ { |
proc setup_md { } { MD++ { |
||
#MD settings |
#MD settings |
||
| Line 198: | Line 192: | ||
MD++ sleep quit |
MD++ sleep quit |
||
</pre> |
</pre> |
||
Instead of creating |
Instead of creating a perfect crystal as the initial condition, this script uses the command '''readcn''' to read the file <tt>300K_5X5X5.cn</tt> created at the end of the previous simulation (NVE ensemble simulation). Note that in this case, the new simulation must use the same time step as the previous simulation that created the .cn file. This is because in MD++ the velocity information are stored as scaled velocity time the time step. If you really need to change the time step in the new simulation (not recommended), you will need to rescale your velocity using the MD++ command '''multiplyvelocity'''. |
||
Setting '''ensemble_type = "NVT"''' activates Nose-Hoover thermostat, which |
Setting '''ensemble_type = "NVT"''' activates the Nose-Hoover thermostat, which adds a variable (<math>\zeta</math>) to the equation of motion. This variable mimicks the heat deposited to the heat reservoir. The coupled equation of motion is |
||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math> \begin{array}{rcl} \ddot\mathbf{r}_i &=& -\frac{1}{m_i}\frac{\partial U(\{\mathbf{r}_i\})}{\partial \mathbf{r}_i} - \zeta \dot\mathbf{r}_i \\ \dot\zeta &=& \frac{3N k_B}{Q} ( T_{\mathrm{inst}} - T_{\mathrm{OBJ}} ) \end{array}</math>. |
|<math> \begin{array}{rcl} \ddot\mathbf{r}_i &=& -\frac{1}{m_i}\frac{\partial U(\{\mathbf{r}_i\})}{\partial \mathbf{r}_i} - \zeta \dot\mathbf{r}_i \\ \dot\zeta &=& \frac{3N k_B}{Q} ( T_{\mathrm{inst}} - T_{\mathrm{OBJ}} ) \end{array}</math>. |
||
|} |
|} |
||
for MD simulations of N particles in 3-dimensions. We can use three different implementations of the Nose-Hoover thermostat in conjunction with the velocity Verlet integrator. When <tt>implementation_type = 0</tt>, an implicit integrator is used. When <tt>implementation_type = 1</tt>, an explicit integrator based on Stormer-Verlet method is used. When <tt>implementation_type = 2</tt>, an explicit integrator baed on Liouville formulation is used. |
|||
when the degree of freedom is 3''N''. |
|||
In MD++, Nose-Hoover thermostat are implemented in three different way, each of which uses different integrator defined through '''implementation_type'''. If it is zero, the implicit integrator will be chosen. If it is one, the explicit integrator based on Sto¨rmer-Verlet method will work. If it is two, another explicit integrator based on Liouville formulation will be activated. |
|||
The MD++ variable '''NHMass''', in the unit of eV·ps<sup>2</sup>, corresponds to the Nose-Hoover thermal mass <math>Q</math> in the above equation of motion. The thermal mass <math>Q</math> can also be specified through the MD++ variable '''vt2''' , in the unit of (1/sec)<sup>2</sup>, by the following relationship |
|||
Thermal coefficient '''vt2''' is related with thermal mass ''Q'' of Nose´-Hoover thermostat as |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math> Q = \frac{3 N k_B T_{\mathrm{OBJ}}}{\mathrm{vt2}}\times (1e+24)</math> |
|<math> Q = \frac{3 N k_B T_{\mathrm{OBJ}}}{\mathrm{vt2}}\times (1e+24)</math>, |
||
|} |
|} |
||
where ''N'' is number of atoms and ''k_B'' is the Boltzmann constant (8.617343e-5 eV/K). |
where ''N'' is number of atoms and ''k_B'' is the Boltzmann constant (8.617343e-5 eV/K). |
||
<!-- For convenience, we also defined thermal mass '''NHMass''', which is exactly the Nose'-Hoover thermal mass ''Q''. --> |
|||
The effect of '''vt2''' on Helmholtz free energy and temperature fluctuation is shown in Fig. 3. |
|||
When '''NHMass''' is non-zero, '''vt2''' will be superceded. When ''N'' = 250 and ''T'' = 300 K (or k_B T ~ 0.026 eV), '''vt2''' = 1e28 (1/sec)<sup>2</sup> corresponds to '''NHMass''' = 1.95e-03 (eV·ps<sup>2</sup>). In comparison with other MD programs such as [http://lammps.sandia.gov LAMMPS], '''vt2''' is proportional to the square of the thermal damping frequency ω, or '''vt2'''~ω<sup>2</sup>. The proportional constant is determined by equating both units. If ω is given in the unit of 1/ps, then the equality becomes '''vt2'''= 10<sup>24</sup>ω<sup>2</sup>. In this case, ω = 100 (1/ps) for '''vt2'''=10<sup>28</sup> (1/sec)<sup>2</sup>. |
|||
The bigger '''vt2''' is (or the smaller '''NHMass''' is), the faster the instantaneous temperature fluctuates around the desired temperature, as shown in Fig. 3. The MD++ simulation also writes out the value of a variable '''HELMP''' in the property file; '''HELMP''' is the total energy of the system plus that of the thermal reservior, and should be conserved. The flucutation of '''HELMP''' around its mean value is also plotted in Fig. 3. |
|||
<gallery caption="Fig.3 The Effect of Nose-Hoover Mass in NVT MD" widths="300px" heights="200px" perrow="2"> |
<gallery caption="Fig.3 The Effect of Nose-Hoover Mass in NVT MD" widths="300px" heights="200px" perrow="2"> |
||
| Line 219: | Line 216: | ||
</gallery> |
</gallery> |
||
In figure 3, you may notice that the energy and the temperature are symmetric before and after the simulation step = 2000. In the above example script, after the first MD simulation (or after the 1st 2000 steps), we use the command '''multiplyvelocity''' to reverse the velocities of all atoms. This is done by setting <tt>input = -1</tt>, so that -1 can be multiplied to the velocities of all atoms and also to ζ. We then continue the MD simulation for the same duration of time as the first one, in order to test the reversibility of the numerical (symplectic) integrator. After the second MD simulation (in the reverse direction), we save the atomic configuration into file <tt>300K-5x5x5-NVT.cn</tt>. Comparing this file with the file <tt>300K-5x5x5.cn</tt>, which contains the atomic configuration at the beginning of the (forward) simulation, reveals that the scaled coordinates of all atoms are identical in the two files within 1e-14 (close to the machine precision). This demonstrates the reversibility of the chosen integrator. If on the other hand, <tt>integrator_type = Gear6</tt> is used, the discrepancy in the atomic coordinates will be on the order of 1e-6, which shows that the Gear6 integrator is not reversible. |
|||
'''HELMP''' in the '''output_fmt''' is Helmholtz free energy which is the conserved quantity in NVT MD. |
|||
'''multiplyvelocity''' multiplies the number specified by the preceding input to all velocity components of all atoms. With '''input=-1''', the MD++ command '''multiplyvelocity''' reverses the velocity direction of all the atoms and they are expected to be at the initial position after the reverse run. This is done to test the reversibility of the symplectic integrator. Comparing two configuration files '''300K-5X5X5.cn''' and '''300K-5X5X5-NVT.cn''' reveals that the numbers become different after 10^(-14) digit. If we consider the total step is 4000, the difference at each step would be ''O''(10^(-17)), which corresponds to the machine precision. On the other hand, if you test '''integrator_type = Gear6''', the numbers become different only after 10^(-6) digit. We can say the symplectic integrator is indeed reversible. |
|||
=== Canonical(NVT) Ensemble with Nose-Hoover Chain === |
=== Canonical(NVT) Ensemble with Nose-Hoover Chain === |
||
---- |
---- |
||
Martyna ''et al''<ref>G. J. Martyna, M. L. Klein and M. Tuckerman, "Nose-Hoover chains: The canonical ensemble via continuous dynamics", J. Chem. Phys. '''97''' 2635--2643</ref> modified the original Nose-Hoover thermostat |
Martyna ''et al''<ref>G. J. Martyna, M. L. Klein and M. Tuckerman, "Nose-Hoover chains: The canonical ensemble via continuous dynamics", J. Chem. Phys. '''97''' 2635--2643</ref> modified the original Nose-Hoover thermostat so that a chain of thermostats are used instead of just one thermostat. The purpose of this chain method is to increase the size of the phase space and help the system to be ergodic when the system is stiff or is not ergodic. Our implementation of the Nose-Hoover chain method in MD++ follows another paper of Martyna.<ref>G. J. Martyna, "Explicit reversible integrators for extended systems dynamics", Molecular Physics '''87''' 1117-1157 (1996)</ref> The following example script, <tt>Mo_NVTC.tcl</tt>, demonstrates how to use the Nose-Hoover chain method in MD++. |
||
The MD++ script '''Mo_NVTC.tcl''', given below, shows how to run Nose-Hoover chain method in MD++ to obtain a system of canonical ensemble. |
|||
<pre> |
<pre> |
||
| Line 236: | Line 229: | ||
# Definition of procedures |
# Definition of procedures |
||
#******************************************* |
#******************************************* |
||
# |
|||
proc initmd { } { MD++ { |
|||
# ... omitted here to save space ... |
|||
.... |
|||
# |
|||
}} |
|||
#------------------------------------------------------------ |
|||
proc readMoPot { } { MD++ { |
|||
.... |
|||
}} |
|||
#------------------------------------------------------------- |
|||
proc openwindow { } { MD++ { |
|||
.... |
|||
}} |
|||
#-------------------------------------------- |
#-------------------------------------------- |
||
proc setup_md { } { MD++ { |
proc setup_md { } { MD++ { |
||
| Line 281: | Line 266: | ||
MD++ run eval |
MD++ run eval |
||
MD++ sleep quit |
MD++ sleep quit |
||
}} |
|||
</pre> |
</pre> |
||
You choose the ensemble type to be '''NVTC'''. The number of chains and masses of each chain can be specified using '''NHChainLen''' and '''NHMass'''. |
|||
According to my experience, '''NHMass''' needs to be chosen with caution, otherwise the system may lose its reversibility, even though Martyna ''et al'' stated that the choice of thermal mass '''NHMass''' is not critical. In fact, they also presented how to take reasonable choice of '''NHMass'''. |
|||
NHMass[0] ~ <math> N_f k_B T / \omega^2</math> where <math>N_f = 3\times N</math> is the number of degrees of freedom. |
|||
And NHMass[i] ~ <math> k_B T / \omega^2</math> (for ''i'' = 2 to ''M'', where ''M'' is the number of chains) |
|||
The '''ensemble_type''' is set to <tt>NVTC</tt> instead of <tt>NVT</tt>. The number of chains is specified in '''NHChainLen''' and the masses for each thermostat on the chain are specified as an array in '''NHMass'''. '''NHMass''' need to be chosen with caution, otherwise the system may lose reversibility, even though Martyna ''et al'' stated that the choice of thermal mass '''NHMass''' is not critical. Martyna ''et all'' suggested the following choice for '''NHMass'''. For a chain of length M that is designed to act as a thermostat for N particles (with <math>N_f = 3 N</math> degrees of freedom), we can set |
|||
thermostat 1 to M -1 fluctuates at a frequency of omega. |
|||
NHMass[0] ~ <math> N_f k_B T / \omega^2</math> |
|||
NHMass[i] ~ <math> k_B T / \omega^2</math> (for ''i'' = 1 to ''M-1'') |
|||
The thermostats <math> i = 1, \cdots, M-1</math> fluctuates with a characteristic frequency <math>\omega</math>. |
|||
=== Constant Stress (N<math>\sigma</math>H) Ensemble === |
=== Constant Stress (N<math>\sigma</math>H) Ensemble === |
||
---- |
---- |
||
In constant stress ensemble, the stress is controlled to |
In MD simulations using a constant stress (N<math>\sigma</math>H) ensemble, the internal Virial stress is controlled to fluctuate around an externally specified stress (i.e. applied stress). In MD++ the Parrinello-Rahman method is implemented. The applied stress tensor <math>\sigma_{\mathrm{EXT}}</math> can be decomposed into a hydrostatic term and a deviatoric term, |
||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 300: | Line 286: | ||
|} |
|} |
||
where <math>\sigma_{\mathrm{EXT}}^{\mathrm{hydro}} = \frac{1}{3}(\sigma_{\mathrm{EXT}})_{kk}\cdot\mathrm{I}</math> and <math>\sigma_{\mathrm{EXT}}^{\mathrm{devi}} = \sigma_{\mathrm{EXT}} - \sigma_{\mathrm{EXT}}^{\mathrm{hydro}}</math>. Here we used the Einstein's |
where <math>\sigma_{\mathrm{EXT}}^{\mathrm{hydro}} = \frac{1}{3}(\sigma_{\mathrm{EXT}})_{kk}\cdot\mathrm{I}</math> and <math>\sigma_{\mathrm{EXT}}^{\mathrm{devi}} = \sigma_{\mathrm{EXT}} - \sigma_{\mathrm{EXT}}^{\mathrm{hydro}}</math>. Here we used the Einstein's notation, in which repeated indices are summed over from 1 to 3, to express the sum of the diagonal components of the stress tensor. The equations of motion of Parrinello-Rahman's method can be written as |
||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 310: | Line 296: | ||
\end{array}</math>. |
\end{array}</math>. |
||
|} |
|} |
||
where <math>\mathbf{H}</math> is the |
where <math>\mathbf{H}</math> is the 3 <math>\times</math> 3 matrix representing the simulation box at time ''t'' and <math>\mathbf{H}_0</math> is the matrix corresponding to the simulation box at ''t'' = 0. <math>\mathbf{G} \equiv \mathbf{H}^T\mathbf{H}</math> is called the metric tensor. <math>\Xi \equiv \Omega \mathbf{H}^{-T}</math> and <math>\Xi_0 \equiv \Omega_0 \mathbf{H}_0^{-T}</math> where <math>\Omega \equiv \det \mathbf{H}</math> and <math>\Omega_0 \equiv \det \mathbf{H}_0</math> are the volumes of the simulation box at time ''t'' and at time 0. ''M'' is an artificial parameter, called the "box mass", that controlls how fast the simulation box changes its shape to reduce the difference between internal (Virial) and applied stress. |
||
If we apply the constant pressure <math>P_{\mathrm{EXT}} = |
If we apply the constant pressure <math>P_{\mathrm{EXT}} = \frac{1}{3}\left(\sigma_{\mathrm{EXT}}\right)_{kk}</math> (notice that in MD++ a pressure correspond to positive stress, which is usually the opposite of the sign convention in elasticity theory), the deviatoric part of the external stress is zero and the second equation becomes simply |
||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math> \ddot\mathbf{H} = -\frac{1}{M} \left[ |
|<math> \ddot\mathbf{H} = -\frac{1}{M} \left[ |
||
| Line 318: | Line 304: | ||
</math>. |
</math>. |
||
|} |
|} |
||
In this case the N<math>\sigma</math>H ensemble reduces to the isoenthalpic-isobaric (NPH) ensemble. The following script, '''Mo_NPH.tcl''', provides an example of how to run NPH MD simulations in MD++. |
|||
<pre> |
<pre> |
||
| Line 327: | Line 313: | ||
# Definition of procedures |
# Definition of procedures |
||
#******************************************* |
#******************************************* |
||
# |
|||
proc initmd { } { MD++ { |
|||
# omitted here to save space ... |
|||
# |
|||
}} |
|||
#------------------------------------------------------------ |
|||
proc readMoPot { } { MD++ { |
|||
... |
|||
}} |
|||
#------------------------------------------------------------- |
|||
proc openwindow { } { MD++ { |
|||
... |
|||
}} |
|||
#-------------------------------------------- |
#-------------------------------------------- |
||
proc setup_md { } { MD++ { |
proc setup_md { } { MD++ { |
||
| Line 347: | Line 325: | ||
0 1000 0 |
0 1000 0 |
||
0 0 1000 ] # compression in MPa |
0 0 1000 ] # compression in MPa |
||
fixboxvec = [ 0 1 1 |
|||
1 0 1 |
|||
1 1 0 ] |
|||
T_OBJ = 300 # (in Kelvin) Desired Temperature |
T_OBJ = 300 # (in Kelvin) Desired Temperature |
||
atommass = 95.94 # (in g/mol) |
atommass = 95.94 # (in g/mol) |
||
| Line 379: | Line 357: | ||
#MD++ finalcnfile = Mo-5X5X5-NPH.cn writecn eval |
#MD++ finalcnfile = Mo-5X5X5-NPH.cn writecn eval |
||
MD++ sleep quit |
MD++ sleep quit |
||
}} |
|||
</pre> |
</pre> |
||
In function <tt>setup_md</tt>, '''ensemble_type''' to set to '''NPH'''. The variable '''wallmass''' is the box mass parameter ''M'' in the Parrinello-Rahman (PR) method. An artificial paramter, '''boxdamp''', (not in the above equation of motion), is introduced to make the system converge faster to an equilibrium state. The effect of '''boxdamp''' is illustrated in Fig. 4. When '''boxdamp''' is nonzero, an additional term is introduced to the box acceleration, as in, |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 389: | Line 367: | ||
|} |
|} |
||
In this case, the dynamics of the system is no longer symplectic (or reversible). The variable '''stress''' is the external stress in unit of MPa. <font color="blue">Note that the sign convention of '''stress''' is opposite of that in the elasticity theory. In MD++ a pressure corresponds to positive normal stresses.</font> '''fixboxvec''' specifies which component of the box matrix <math>\mathrm{H}</math> will be fixed during the MD simulation; the corresponding matrix element in '''fixboxvec''' is 1. |
|||
you need to specify <math>\mathrm{H}_0</math> by calling '''saveH''' before using the PR method. According to Ray and Rahman<ref>J. R. Ray and A. Rahman, "Statistical ensembles and molecular dynamics studies of anisotropic solids", J. Chem. Phys, '''80''' 4423--4428 </ref>, "<math>\mathrm{H}_0</math> should be taken as the average value of <math>\mathrm{H}</math> when the stress is zero." |
|||
The command '''saveH''' stores the current box matrix H into <math>\mathrm{H}_0</math> in the Parrinello-Rahman method. This needs to be done before starting the MD simulation. According to Ray and Rahman<ref>J. R. Ray and A. Rahman, "Statistical ensembles and molecular dynamics studies of anisotropic solids", J. Chem. Phys, '''80''' 4423--4428 </ref>, <math>\mathrm{H}_0</math> should be taken as the average value of <math>\mathrm{H}</math> when the stress is zero. |
|||
<gallery caption="Fig.4 The Effect of |
<gallery caption="Fig.4 The Effect of BoxDamp paramter in NPH MD simulation. A finite BoxDamp speeds up the convergence toward an equilibrium state." widths="250px" heights="200px" perrow="3"> |
||
Image:Ext_energy_fluct_NPH.jpg|(a) |
Image:Ext_energy_fluct_NPH.jpg|(a) Fluctuation of the total energy of the extended system (energy of the original system plus that of the box) -- a conserved quantity, as a function of time. |
||
Image:Stress_fluct_NPH.jpg|(b) Stress in NPH ensemble. |
Image:Stress_fluct_NPH.jpg|(b) Stress fluctuation in NPH ensemble. |
||
Image:Box_fluct.jpg|(c) |
Image:Box_fluct.jpg|(c) Size of the simulation box as a function of time. |
||
</gallery> |
</gallery> |
||
Latest revision as of 17:26, 20 January 2011
Manual 05 for MD++
Molecular Dynamics Simulations
Keonwook Kang and Wei Cai
[First written, [ Jan 19 ]], 2007
[Last Modified, [ Apr 13 ]], 2008
The case studies described in Manuals 02-04 only deal with atomic positions as degrees of freedom. For example, in Manual 02, we learned how to create a perfect crystal by MD++ command makecrystal. In Manual 03, we varied the size of the perfect crystal to find the equilibrium lattice constants. In Manual 04, we introduced a vacancy to the perfect crystal and let the atomic positions relax to a local energy minimum. In this manual, we will learn how to perform finite temperature, Molecular Dynamics (MD) simulations. For this purpose, we will need to deal with atomic velocities as degrees of freedom, as well as a number of control variables, such as integrator type, time step, and output controls. We will learn to specify the simulation ensembles from different choices: microcanonical (NVE), canonical (NVT), isoenthalpic-isobaric (NPH) and constant pressure/temperature (NPT) ensembles. Different quantities are conserved or controlled in different ensembles. For example, the total number of atoms (N), volume (V) and energy (E) are conserved in the (NVE) ensemble.
Microcanonical (NVE) Ensemble
The NVE ensemble is the probably the best starting point to understand what happens in MD simulations. In Statistical Mechanics, to picture the NVE ensemble, we can imagine a system of N gas molecules inside a rigid container with perfect thermal insulation. Hence no work or heat can be exchanged between the system and the outside world (its environment). While the gas molecules move with time, the total number of particles N, the total energy E and the total volume V of the system does not change. A large number of replicas of the system with the same N, V, E forms the NVE, or microcanonical ensemble in Statistical Mechanics. In MD simulations within the NVE ensemble, we usually do not simulate the rigid container --- this is because the simulation volume is usually very small and having an explicit container will lead to a large surface-to-volume ratio and large simulation artifacts. Instead, periodic boundary conditions (PBC) are usually used in MD simulation. If the repeat vectors of PBC do not change, the simulation volume V is kept constant during the simulation. Since the particles follows Hamiltonian dynamics in MD simulations, the total energy E is conserved. The total number of particles N is obviously conserved in the MD simulation. Therefore, MD simulations under PBC with fixed repeat vectors and no thermostats (to be described later) correspond to the NVE (microcanonical) ensemble.
The following input script mo_NVE.tcl gives an example of running MD simulations in MD++. You can test is by the following command (if you put this file in your scripts/ directory)
$ bin/fs_gpp scripts/mo_NVE.tcl
This example script first creates a perfect BCC crystal of molybdenum (Mo) with a supercell of 5[1 0 0] Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times} 5[0 1 0] Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times} 5[0 0 1]. It then runs MD simulations using the Finnis-Sinclair (FS) potential.
# -*-shell-script -*-
# run NVE MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
proc initmd { } { MD++ {
#setnolog
setoverwrite
dirname = runs/mo-example
zipfiles = 1 # zip output files
#
#--------------------------------------------
# Create Perfect Crystal
element0 = Mo
crystalstructure = body-centered-cubic
latticeconst = 3.1472 # in Angstrom for Mo
latticesize = [ 1 0 0 5
0 1 0 5
0 0 1 5]
}}
#------------------------------------------------------------
proc readMoPot { } { MD++ {
# Read the potential file
potfile = ~/Codes/MD++/potentials/mo_pot readpot
}}
#-------------------------------------------------------------
proc openwindow { } { MD++ {
# Plot Configuration
atomradius = 1.0 bondradius = 0.3 bondlength = 2.725
atomcolor = orange highlightcolor = purple
bondcolor = red backgroundcolor = gray70
plotfreq = 10 rotateangles = [ 0 0 0 1.25 ]
openwin alloccolors rotate saverot eval plot
}}
#--------------------------------------------
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NVE" integrator_type = "VVerlet"
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (in g/mol)
timestep = 0.001 # (in ps)
totalsteps = 2000
# save property every 10 steps
saveprop = 1 savepropfreq = 10
savecn = 0 savecnfreq = 100
writeall = 1 DOUBLE_T = 1
randseed = 12345 srand48 #randomize random number generator
}}
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ makecrystal finalcnfile = perf.cn writecn
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ initvelocity finalcnfile = init.cn writecn
#format of thermodynamic property file
MD++ {output_fmt = "curstep EPOT KATOM Tinst"}
MD++ outpropfile = thermo.out openpropfile
MD++ run closepropfile
MD++ finalcnfile = 300K-5X5X5.cn writecn eval
MD++ sleep quit
In this Tcl script, we start with some definition of functions that will be used subsequently. The main program starts after the comment Main program starts here. The program first calls the initmd function that was defined at the beginning, which opens the output directory. After creating the perfect crystal using the makecrystal command, it calls the setup_md function that initializes some important parameters for MD simulation. (Commands and variables related with visualization as specified in the openwindow function will be covered in a separate manual in detail.)
The first line in the setup_md function selects the statistica ensemble for the simulation. The possible choices for the variable ensemble_type are NVE, NVT, NPH and NPT. The type of the numerical integrator is specified by the integrator_type variable and can be either the Gear 6-th order predictor-corrector algorithm (Gear6) or the velocity Verlet algorithm (VVerlet). The predictor-corrector integrator has a higher order of accuracy but velocity Verlet is a symplectic integrator and is more stable at larger algorithm. We recommend the use of velocity Verlet algorithm whenever possible (i.e. if it is implemented for the chosen ensemble type).
The variable atommass specifies the atomic mass in unit of g/mol. timestep specifies the integrator time step Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Delta t} in unit of picosecond. totalsteps is the total number of time steps for the MD simulation. Hence totalsteps times timestep is the total time duration of the MD simulation. We usually choose timestep to be as large as possible provided that the total energy of the system is conserved within an acceptable accurady. totalsteps is usually chosen so that the total simulation time is long enough for the system to reach an equilibrium or steady state (usually on the order of picoseconds).
The command initvelocity assign random numbers to the atomic velocities and then scale them so that the instantaneous temperature matches the target value, as specified by T_OBJ in unit of K. Strictly speaking, temperature is not well defined in an NVE ensemble (but is well defined in the NVT ensemble). Here we can regard T_OBJ as a measure of the instantaneous kinetic energy of the system. MD++ uses the drand48() to generate random numbers for the velocities. The random number generator can be initialized by calling srand48 with a specified randseed. This approach is more convenient for debugging because the same randseed garauntees exactly the same random number sequence will be generated if you run the simulation again. On the other hand, you may use the function srandbytime to use the current time as the random seed for initializing the random number generator. This makes sure that ever time you run the simulation again a completely different set of random numbers will be generated.
If the parameter DOUBLE_T is set to 1, then the velocities are scaled in such a way that the instantaneous temperature is twice of T_OBJ, as illustrated in Fig.1. The reason we may want to set DOUBLE_T = 1 is the following. When we assign velocities to the atoms in a perfect crystal, the instantaneous temperature almost always drop to half of its initial value when thermal equilibrium is reached (see Fig.1). This is due to the energy equipartition theorem. For solids at temperatures much below the melting temperature, the Hamiltonian is close to that of a set of coupled harmonic oscillators. The total energy of a harmonic system is equally divided between the average kinetic energy and the average potential energy. Because the total energy is conserved in an NVE-ensemble simulation, the kinetic energy is bound to decrease by half if the kinetic energy and potential energy (initially zero) are equal (in time average) when equilibrium is reached. Setting DOUBLE_T = 1 allows the temperature at the equilibrium state to match the desired temperature.
- Fig.1 Instantaneous temperature as a function of time during MD simulations with different settings for DOUBLE_T.
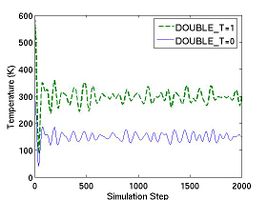

The parameters saveprop and savepropfreq specify whether or not and how often the simulation properties will be saved periodically in a data file. If saveprop = 1 and savepropfreq = 10, the properties such as potential energy, kinetic energy and temperature will be save every 10 simulation steps, provided that the MD++ command openpropfile is called before the command run. The name of the property file is specified by the outputfile variable and is prop.out by default. The setting zipfiles = 1 at the beginning of this script file (line 11) specifies that both property files and atomic configuration files (see below) will be automatically zipped (by gzip) after they are written.
The content of a property file is defined by variable output_fmt. In this example, each line of the property file will contain the current step (curstep), potential energy (EPOT), kinetic energy (KATOM), and temperature (Tinst). If we do not specify output_fmt, the default content of the property file will be current step (curstep), kinetic energy (KATOM), potential energy (EPOT), pressure (PRESSURE), Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{xy}} (TSTRESS_xy), Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{yz}} (TSTRESS_yz), Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{zx}} (TSTRESS_zx), HELM, the extended energy(HELMP), thermodynamic friction coefficient (zeta), reversible work increment dEdlambda and volume (OMEGA). The energies are in unit of eV and stresses in eV/A˚^3. Please note that there is an overall minus sign between the stress variables in MD++ and that in conventional elasticity theory. For example, a positive pressure correspond to positive normal stress components in MD++ but negative normal stress components in elasticity theory. This is because in MD++, we followed the sign convention of Parrinello and Rahman for stress control.[1]
The parameters savecn and savecnfreq specify whether or not and how often the atomic coordinates (and velocities if writeall = 1) will be saved as .cn files during the MD simulation. If savecn = 1 and savepropfreq = 10000, the intermediate .cn files will be saved every 10000 steps, provided that the MD++ command openintercnfile is called before the command run. The name of the intermediate .cn files are specified by the intercnfile variable and is inter####.cn by default, where #### are integers (starting with 0000) that will be automatically incremented by one after each file is written. In this example, savecn = 0 so no intermediate .cn files will be saved. After all the relevant parameters have been set up, the MD++ command run starts the MD simulation.
After the simulation is finished, a property file named thermo.out will be written in the output directory runs/mo-example. This file can be loaded and plotted by programs such as Matlab, Octave, or Gnuplot.[2] Fig.2 plots the total energy per atom and instantanous temperature for two simulations with different sizes. The larger system (10[100] 10[010] 10[001]) has eight times more atoms than the smaller one (5[100] Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times} 5[010] Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times} 5[001]). As expected, the data for the larger system experience less statistical fluctuation.

- Fig.2 Energy and Temperature fluctuation of NVE ensemble
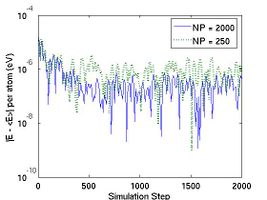
(a) Energy Fluctuation in NVE ensemble.
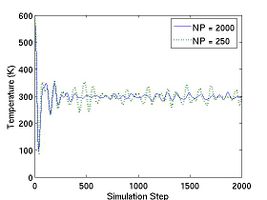
(b) Temperature in NVE ensemble


After the run command, the script file calls the command writecn to save the final atomic configuration into a .cn file whose name is specified by finalcnfile. The .cn file has the following format. The first line contains the total number of atoms NP. The following NP lines then contain the scaled coordinates of all atoms: Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle s_x^{(i)}}
, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle s_y^{(i)}}
, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle s_z^{(i)}}
, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle i = 1, 2, \cdots NP}
. The last three lines of the .cn file specify a 3 Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times}
3 matrix Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}}
whose column vectors are the three repeat vectors of the simulation supercell (subjected to periodic boundary conditions). The real coordinates of an atom (x, y, z ) and the scaled coordinates for each atom are related through Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}}
as follows
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \left( \begin{array}{c} x \\ y \\ z \end{array} \right) = \left( \begin{array}{ccc} H_{11} & H_{12} & H_{13} \\ H_{21} & H_{22} & H_{23} \\ H_{31} & H_{32} & H_{33} \end{array} \right) \left( \begin{array}{c} s_x \\ s_y \\ s_z \end{array} \right)} . |
If writeall = 1 is specified before writecn, additional informations are added to the NP lines of data. Following the scaled coordinates, each line will also contain the scaled velocities (_VSR[i].x, _VSR[i].y and _VSR[i].z), local potential energy (_EPOT_IND[i]), flag (fixed[i]), central-symmetry-deviation paramter (_TOPOL[i]), atom species (species[i]), atom groups (group[i]) and image index (image[i]). You can see this difference by comparing two configuration files, perf.cn (written before setting writeall = 1) and init.cn (written after). If writevelocity = 1 is used instead of writeall = 1, then only the scaled velocities are added behind the three columns for scaled coordinates.
At the end of the example script, the command sleep is called so that the graphics window to stay open (i.e. alive) for a while so that we can examine the atomic structure. You can press ctrl-c in the terminal to exit. You can also comment out the sleep command in the script file so that MD++ exits immediately after reaching the end of the input file.
Canonical(NVT) Ensemble
In the NVT ensemble of Statistical Mechanics, the system exchanges heat with an external thermostat to maintain its temperature at T but its total energy is no longer conserved. The following script mo_NVT.tcl provides an example of NVT simulations in MD++. You can test it by typing
$ bin/fs_gpp scripts/mo_NVT.tcl
This script first reads in an atomic configuration generated by the previous MD simulation (NVE ensemble) and then performs MD simulation in the NVT ensemble at T = 300 K using the Nose-Hoover thermostat[3].
# -*-shell-script -*-
# run NVT MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
#
# ... omitted here to save space ...
#
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NVT" integrator_type = "VVerlet"
implementation_type = 0 vt2 = 1e28
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (g/mol)
timestep = 0.001 # (ps)
totalsteps = 2000
saveprop = 1 savepropfreq = 10
savecn = 0 savecnfreq = 100
writeall = 1
}}
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ {
incnfile = 300K-5X5X5.cn
atommass = 95.94 timestep = 0.001
readcn eval
}
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ {output_fmt = "curstep EPOT KATOM Tinst HELMP"}
MD++ outpropfile = thermo-NVT.out openpropfile
MD++ run eval
#reverse velocity (to test reversibility)
MD++ input = -1 multiplyvelocity
#MD simulation (reverse)
MD++ run eval
MD++ finalcnfile = 300K-5X5X5-NVT.cn writecn
MD++ sleep quit
Instead of creating a perfect crystal as the initial condition, this script uses the command readcn to read the file 300K_5X5X5.cn created at the end of the previous simulation (NVE ensemble simulation). Note that in this case, the new simulation must use the same time step as the previous simulation that created the .cn file. This is because in MD++ the velocity information are stored as scaled velocity time the time step. If you really need to change the time step in the new simulation (not recommended), you will need to rescale your velocity using the MD++ command multiplyvelocity.
Setting ensemble_type = "NVT" activates the Nose-Hoover thermostat, which adds a variable (Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \zeta} ) to the equation of motion. This variable mimicks the heat deposited to the heat reservoir. The coupled equation of motion is
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \begin{array}{rcl} \ddot\mathbf{r}_i &=& -\frac{1}{m_i}\frac{\partial U(\{\mathbf{r}_i\})}{\partial \mathbf{r}_i} - \zeta \dot\mathbf{r}_i \\ \dot\zeta &=& \frac{3N k_B}{Q} ( T_{\mathrm{inst}} - T_{\mathrm{OBJ}} ) \end{array}} . |
for MD simulations of N particles in 3-dimensions. We can use three different implementations of the Nose-Hoover thermostat in conjunction with the velocity Verlet integrator. When implementation_type = 0, an implicit integrator is used. When implementation_type = 1, an explicit integrator based on Stormer-Verlet method is used. When implementation_type = 2, an explicit integrator baed on Liouville formulation is used.
The MD++ variable NHMass, in the unit of eV·ps2, corresponds to the Nose-Hoover thermal mass Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle Q} in the above equation of motion. The thermal mass Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle Q} can also be specified through the MD++ variable vt2 , in the unit of (1/sec)2, by the following relationship
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle Q = \frac{3 N k_B T_{\mathrm{OBJ}}}{\mathrm{vt2}}\times (1e+24)} , |
where N is number of atoms and k_B is the Boltzmann constant (8.617343e-5 eV/K). When NHMass is non-zero, vt2 will be superceded. When N = 250 and T = 300 K (or k_B T ~ 0.026 eV), vt2 = 1e28 (1/sec)2 corresponds to NHMass = 1.95e-03 (eV·ps2). In comparison with other MD programs such as LAMMPS, vt2 is proportional to the square of the thermal damping frequency ω, or vt2~ω2. The proportional constant is determined by equating both units. If ω is given in the unit of 1/ps, then the equality becomes vt2= 1024ω2. In this case, ω = 100 (1/ps) for vt2=1028 (1/sec)2.
The bigger vt2 is (or the smaller NHMass is), the faster the instantaneous temperature fluctuates around the desired temperature, as shown in Fig. 3. The MD++ simulation also writes out the value of a variable HELMP in the property file; HELMP is the total energy of the system plus that of the thermal reservior, and should be conserved. The flucutation of HELMP around its mean value is also plotted in Fig. 3.
- Fig.3 The Effect of Nose-Hoover Mass in NVT MD
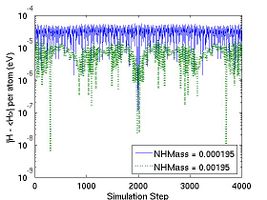
(a) Helmholtz energy fluctuation in NVT ensemble.
(b) Temperature in NVT ensemble


In figure 3, you may notice that the energy and the temperature are symmetric before and after the simulation step = 2000. In the above example script, after the first MD simulation (or after the 1st 2000 steps), we use the command multiplyvelocity to reverse the velocities of all atoms. This is done by setting input = -1, so that -1 can be multiplied to the velocities of all atoms and also to ζ. We then continue the MD simulation for the same duration of time as the first one, in order to test the reversibility of the numerical (symplectic) integrator. After the second MD simulation (in the reverse direction), we save the atomic configuration into file 300K-5x5x5-NVT.cn. Comparing this file with the file 300K-5x5x5.cn, which contains the atomic configuration at the beginning of the (forward) simulation, reveals that the scaled coordinates of all atoms are identical in the two files within 1e-14 (close to the machine precision). This demonstrates the reversibility of the chosen integrator. If on the other hand, integrator_type = Gear6 is used, the discrepancy in the atomic coordinates will be on the order of 1e-6, which shows that the Gear6 integrator is not reversible.
Canonical(NVT) Ensemble with Nose-Hoover Chain
Martyna et al[4] modified the original Nose-Hoover thermostat so that a chain of thermostats are used instead of just one thermostat. The purpose of this chain method is to increase the size of the phase space and help the system to be ergodic when the system is stiff or is not ergodic. Our implementation of the Nose-Hoover chain method in MD++ follows another paper of Martyna.[5] The following example script, Mo_NVTC.tcl, demonstrates how to use the Nose-Hoover chain method in MD++.
# -*-shell-script -*-
# run NVE MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
#
# ... omitted here to save space ...
#
#--------------------------------------------
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NVTC" integrator_type = "VVerlet"
NHChainLen = 4 # MAXNHCLEN = 20 in md.h
NHMass = [ 1.95e-4 2e-6 2e-6 2e-6 ]
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (in g/mol)
timestep = 0.001 # (in ps)
totalsteps = 5000
saveprop = 1 savepropfreq = 10
savecn = 0 savecnfreq = 100
writeall = 1
DOUBLE_T = 0 randseed = 12345 srand48
}}
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ {
incnfile = ../mo-example/300K-5X5X5.cn
atommass = 95.94 timestep = 0.001
readcn eval
}
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ {output_fmt = "curstep EPOT KATOM Tinst HELMP"}
MD++ outpropfile = thermo-NVTC.out openpropfile
MD++ run eval
MD++ sleep quit
The ensemble_type is set to NVTC instead of NVT. The number of chains is specified in NHChainLen and the masses for each thermostat on the chain are specified as an array in NHMass. NHMass need to be chosen with caution, otherwise the system may lose reversibility, even though Martyna et al stated that the choice of thermal mass NHMass is not critical. Martyna et all suggested the following choice for NHMass. For a chain of length M that is designed to act as a thermostat for N particles (with Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle N_f = 3 N}
degrees of freedom), we can set
NHMass[0] ~ Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle N_f k_B T / \omega^2}
NHMass[i] ~ Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_B T / \omega^2}
(for i = 1 to M-1)
The thermostats Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle i = 1, \cdots, M-1} fluctuates with a characteristic frequency Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \omega} .
Constant Stress (NFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma} H) Ensemble
In MD simulations using a constant stress (NFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma} H) ensemble, the internal Virial stress is controlled to fluctuate around an externally specified stress (i.e. applied stress). In MD++ the Parrinello-Rahman method is implemented. The applied stress tensor Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{\mathrm{EXT}}} can be decomposed into a hydrostatic term and a deviatoric term,
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{\mathrm{EXT}} = \sigma_{\mathrm{EXT}}^{\mathrm{hydro}} + \sigma_{\mathrm{EXT}}^{\mathrm{devi}} } . |
where Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{\mathrm{EXT}}^{\mathrm{hydro}} = \frac{1}{3}(\sigma_{\mathrm{EXT}})_{kk}\cdot\mathrm{I}} and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{\mathrm{EXT}}^{\mathrm{devi}} = \sigma_{\mathrm{EXT}} - \sigma_{\mathrm{EXT}}^{\mathrm{hydro}}} . Here we used the Einstein's notation, in which repeated indices are summed over from 1 to 3, to express the sum of the diagonal components of the stress tensor. The equations of motion of Parrinello-Rahman's method can be written as
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \begin{array}{rcl} \ddot\mathbf{r}_i &=& -\frac{1}{m_i}\frac{\partial U(\{\mathbf{r}_i\})}{\partial \mathbf{r}_i} - \mathbf{H}^{-T}\dot\mathbf{G}\mathbf{H}^{-1} \dot\mathbf{r}_i \\ \ddot\mathbf{H} &=& -\frac{1}{M} \left[ \left( \sigma_{\mathrm{Virial}} - \sigma_{\mathrm{EXT}}^{\mathrm{hydro}} \right)\Xi - \left( \mathbf{H}\mathbf{H}_0^{-1} \sigma_{\mathrm{EXT}}^{\mathrm{devi}} \right) \Xi_0 \right] \end{array}} . |
where Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}} is the 3 Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \times} 3 matrix representing the simulation box at time t and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}_0} is the matrix corresponding to the simulation box at t = 0. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{G} \equiv \mathbf{H}^T\mathbf{H}} is called the metric tensor. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Xi \equiv \Omega \mathbf{H}^{-T}} and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Xi_0 \equiv \Omega_0 \mathbf{H}_0^{-T}} where Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Omega \equiv \det \mathbf{H}} and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Omega_0 \equiv \det \mathbf{H}_0} are the volumes of the simulation box at time t and at time 0. M is an artificial parameter, called the "box mass", that controlls how fast the simulation box changes its shape to reduce the difference between internal (Virial) and applied stress.
If we apply the constant pressure Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle P_{\mathrm{EXT}} = \frac{1}{3}\left(\sigma_{\mathrm{EXT}}\right)_{kk}} (notice that in MD++ a pressure correspond to positive stress, which is usually the opposite of the sign convention in elasticity theory), the deviatoric part of the external stress is zero and the second equation becomes simply
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \ddot\mathbf{H} = -\frac{1}{M} \left[ \left( \sigma_{\mathrm{Virial}} + P_{\mathrm{EXT}}\mathbf{I} \right)\Xi \right] } . |
In this case the NFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma} H ensemble reduces to the isoenthalpic-isobaric (NPH) ensemble. The following script, Mo_NPH.tcl, provides an example of how to run NPH MD simulations in MD++.
# -*-shell-script -*-
# run NPH MD simulation of perfect crystal of Mo.
#
#*******************************************
# Definition of procedures
#*******************************************
#
# omitted here to save space ...
#
#--------------------------------------------
proc setup_md { } { MD++ {
#MD settings
ensemble_type = "NPH" integrator_type = "VVerlet"
wallmass = 1000 boxdamp = 0.1
saveH # Use current H as reference (H0), needed for specifying stress
stress = [ 1000 0 0
0 1000 0
0 0 1000 ] # compression in MPa
fixboxvec = [ 0 1 1
1 0 1
1 1 0 ]
T_OBJ = 300 # (in Kelvin) Desired Temperature
atommass = 95.94 # (in g/mol)
timestep = 0.001 # (in ps)
totalsteps = 10000
saveprop = 1 savepropfreq = 20
savecn = 0 savecnfreq = 100
writeall = 1
DOUBLE_T = 0 randseed = 12345 srand48
}}
#*******************************************
# Main program starts here
#*******************************************
initmd
readMoPot
MD++ makecrystal finalcnfile = perf.cn writecn
openwindow
#---------------------------------------------
# run MD
setup_md
MD++ initvelocity finalcnfile = init.cn writecn
MD++ {output_fmt = "curstep EPOT KATOM Tinst HELMP \
TSTRESSinMPa_xx TSTRESSinMPa_yy TSTRESSinMPa_zz \
TSTRESSinMPa_xy TSTRESSinMPa_xz TSTRESSinMPa_yz \
H_11 H_22 H_33 H_12 H_13 H_21 H_23 H_31 H_32" }
MD++ outpropfile = thermo-NPH.out openpropfile
MD++ run closepropfile
#MD++ finalcnfile = Mo-5X5X5-NPH.cn writecn eval
MD++ sleep quit
In function setup_md, ensemble_type to set to NPH. The variable wallmass is the box mass parameter M in the Parrinello-Rahman (PR) method. An artificial paramter, boxdamp, (not in the above equation of motion), is introduced to make the system converge faster to an equilibrium state. The effect of boxdamp is illustrated in Fig. 4. When boxdamp is nonzero, an additional term is introduced to the box acceleration, as in,
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \ddot\mathbf{H} := \ddot\mathbf{H}- (\mathrm{boxdamp}) \times \dot\mathbf{H} } |
In this case, the dynamics of the system is no longer symplectic (or reversible). The variable stress is the external stress in unit of MPa. Note that the sign convention of stress is opposite of that in the elasticity theory. In MD++ a pressure corresponds to positive normal stresses. fixboxvec specifies which component of the box matrix Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathrm{H}} will be fixed during the MD simulation; the corresponding matrix element in fixboxvec is 1.
The command saveH stores the current box matrix H into Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathrm{H}_0} in the Parrinello-Rahman method. This needs to be done before starting the MD simulation. According to Ray and Rahman[6], Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathrm{H}_0} should be taken as the average value of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathrm{H}} when the stress is zero.
- Fig.4 The Effect of BoxDamp paramter in NPH MD simulation. A finite BoxDamp speeds up the convergence toward an equilibrium state.
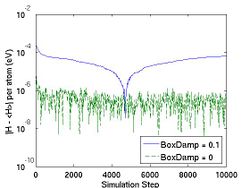
(a) Fluctuation of the total energy of the extended system (energy of the original system plus that of the box) -- a conserved quantity, as a function of time.
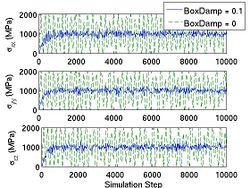
(b) Stress fluctuation in NPH ensemble.
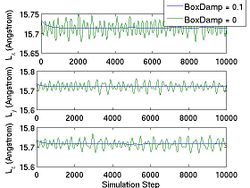
(c) Size of the simulation box as a function of time.



Isothermal-isobaric (NPT) Ensemble
- ↑ M. Parrinello and A. Rahman, "Polymorphic transitions in single crystals: A new molecular dynamics method", J. Appl. Phys 52 7182-7190 1981
- ↑ A simple way to plot the total energy using gnuplot (a free software on Unix/Linux) is to run the command. plot "thermo.out" u ($1):($2+$3) with line
- ↑ S. Nose, “A Molecular Dynamics Method for Simulations in the Canonical Ensemble”, Molecular Physics, 52 255 (1984)
- ↑ G. J. Martyna, M. L. Klein and M. Tuckerman, "Nose-Hoover chains: The canonical ensemble via continuous dynamics", J. Chem. Phys. 97 2635--2643
- ↑ G. J. Martyna, "Explicit reversible integrators for extended systems dynamics", Molecular Physics 87 1117-1157 (1996)
- ↑ J. R. Ray and A. Rahman, "Statistical ensembles and molecular dynamics studies of anisotropic solids", J. Chem. Phys, 80 4423--4428