M09 Computing Ideal Strength: Difference between revisions
| (33 intermediate revisions by 3 users not shown) | |||
| Line 6: | Line 6: | ||
<P ALIGN="CENTER"><STRONG>Keonwook Kang and Wei Cai</STRONG></P> |
<P ALIGN="CENTER"><STRONG>Keonwook Kang and Wei Cai</STRONG></P> |
||
</DIV> |
</DIV> |
||
<P ALIGN="CENTER">[[ |
<P ALIGN="CENTER">[[Dec. 8]], [[2007]]</P> |
||
<P> |
<P> |
||
<BR><HR> |
<BR><HR> |
||
The ideal strength of a material is the maximum stress that it can sustain and is an upper bound to the critical stress for crack and dislocation nucleation in the material. When the applied stress exceeds the ideal strength, the crystal structure will collapse even at zero temperature. The mode of failure depends on the type of applied stress. When the tensile stress exceeds the ideal tensile strength, the crystal is unstable against cleavage fracture. When the shear stress exceeds the ideal shear strength, the crysal is unstable against slip on crystallographic planes. Because a real crystal is not perfect but contains defects (such as surfaces, grain boundaries, dislocations, vacancies), its strength is lower than the ideal strength. When the defect density is made sufficiently small (such as in whiskers), the strength of a real crystal can approach the ideal strength. The ideal strength is also called the ''theoretical'' strength. |
|||
Ideal strength is the theoretical limit up to which a defect-free material can |
|||
sustain external load before it breaks. We define ideal strength as the maximum |
|||
There are two alternative definitions of the ideal strength depending on the way that the deformation is imposed on the perfect crystal structure. In the first definition, the deformation strain can be applied uniformly to the simulation cell (usually under periodic boundary conditions) resulting in an affine deformation of the entire crystal<ref>D. Roudy and M. L. Cohen, Phys. Rev. B '''64''' 212103 (2001))</ref>. In the second definition, the deformation strain can be localized between two crystallographic planes<ref name=kangcai2007>Keonwook Kang and Wei Cai, Philosophical Magazine, '''87''' 2169–2189 (2007)</ref>. The ideal strength values obtained from these two approaches are somewhat different but are also expected to co-relate with each other. In this manual, we describe how to compute the ideal strength according to the second definition, using diamond-cubic structure of Si as an example. |
|||
derivative of the excess energy (per area) that comes from rigid translation of |
|||
part of material.<ref>There is different definition of ideal strength. Roudy and Cohen calculated it from the excess energy of uniform deformation of material.(D. Roudy and M. L. Cohen, Phys. Rev. B '''64''' 212103 (2001))</ref> In this manual, it is explained how the ideal strengths of diamond cubic structure of Si are calculated with MD++ codes. This task was part of work covered in our paper<ref name=kangcai2007>Keonwook Kang and Wei Cai, Philosophical Magazine, '''87''' 2169–2189 (2007)</ref>. |
|||
<BR><HR> |
<BR><HR> |
||
== Ideal Tensile Strength == |
== Ideal Tensile Strength == |
||
Suppose we want to compute the ideal tensile strength of Si along the [1 1 0] direction, i.e., <math>\sigma_{[110]}^{\mathrm{th}}</math>. First, we create a simulation cell under periodic boundary condition (PBC) with the three periodicity vectors <math>\mathbf{c}_1 = |
|||
N_x [1 1 0]</math>, <math>\mathbf{c}_2 = N_y[\bar{1} 1 0]</math> and <math>\mathbf{c}_3 = N_z[0 0 1]</math>. The coordinate system is defined such that <math>\mathbf{c}_1</math>, <math>\mathbf{c}_2</math> and <math>\mathbf{c}_3</math> are aligned along <math>x</math>, <math>y</math> and <math>z</math> directions, respectively. The crystal just created is surrounded by its own images in all three directions. |
|||
First, a simulation cell is created with the three periodicity vectors <math>\mathbf{c}_1 = |
|||
N_x [1 1 0]</math>, <math>\mathbf{c}_2 = N_y[\bar{1} 1 0]</math> and <math>\mathbf{c}_3 = N_z[0 0 1]</math>, each of which is aligned along <math>x</math>, <math>y</math> and <math>z</math> direction, respectively. Make sure that the cell is subjected to periodic |
|||
Next, we change the length of the repeat vector <math>\mathbf{c}_1</math> while keeping the position of all atoms in the simulation cell fixed. Let <math>\mathbf{c}_1^0</math> be the original value of the repeat vector along the <math>x</math> direction and <math>L_x^0 = |\mathbf{c}_1^0|</math>. Suppose we assign the new repeat vector to be |
|||
boundary condition (PBC) in all three directions. Next, the potential energy |
|||
change is recorded as the function of the opening distance <math>d</math> when the (1 1 0) |
|||
surface opens rigidly (See Fig. 1(a).) or |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math> |
|<math>\mathbf{c}_1 = \left( 1 + \frac{d}{L_x^0}\right) \mathbf{c}_1^0 </math>. |
||
|} |
|} |
||
This will create a gap of width <math>d</math> between the crystal in the simulation cell (primary cell) with its images in the <math>x</math> direction, as shown in Fig. 1(a). For each value of <math>d</math>, we compute the potential energy of the crystal (without relaxing the positions of the atoms). The excess energy with respect to the case of <math>d=0</math>, dividied by the cross section area of the simulation cell perpendicular to the <math>x</math> direction, is defined as function of <math>d</math> |
|||
where <math>\gamma</math> is the excess energy to create two free surfaces. In order to rigidly separate a bulk crystal, we could collectively translate half of atoms but instead |
|||
we used a simple trick. When we redefine the periodicity vector <math>\mathbf{c}_1</math> as |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math>\ |
|<math> \gamma = \gamma ( d ) </math>. |
||
|} |
|} |
||
and is plotted in Fig. 2(a). |
|||
while fixing the coordinates of all the atoms, it gives the same effect of separating |
|||
the bulk crystal into two by <math>d</math>. We change the box length <math>L_x \equiv a|\mathbf{c}_1|</math> by redefining the periodicity vector <math>\mathbf{c}_1</math> from its initial definition <math>\mathbf{c}^0_1</math> with certain |
|||
ratio and it will create a gap of width <math>d</math> in <math>x</math> direction between the periodic image (2) and the crystal (1) in the primary cell as shown in Fig. 1(a). |
|||
In MD++, the transformation of the simulation cell as described above can be performed conveniently using the command '''changeH_keepR'''. <math>\mathbf{H}</math> is a 3 by 3 matrix whose three columns correspond to the coordinates of the three repeat vectors, i.e. <math>\mathbf{H} = [\mathbf{c}_1 | \mathbf{c}_2 | \mathbf{c}_3]</math>. The syntax for this command is |
|||
In MD++ Codes, there is a command '''changeH_keepR''' which changes one of |
|||
periodicity vectors with atoms fixed. The input to this command has three |
|||
numbers. |
|||
input = [ i j frac ] changeH_keepR |
input = [ ''i'' ''j'' ''frac'' ] changeH_keepR |
||
This will lead to the following transformation of the repeat vectors |
|||
The first number is the index of the periodicity vector that you want |
|||
to change. For example, if '''i=1''', <math>\mathbf{c}_1</math> vector will be changed. (2 for <math>\mathbf{c}_2</math> and 3 for <math>\mathbf{c}_3</math>) The second number is the index of direction. If '''j=1''', the fraction '''frac''' of the vector <math>\mathbf{c}_1</math> is added to itself and <math>\mathbf{c}_1</math> can be longer or shorter depending on the sign of '''frac'''. If '''j=2''', the vector <math>\mathbf{c}_1</math> will be tilted by the fraction '''frac''' of <math>\mathbf{c}_2</math> vector. |
|||
{|border="0" align="center" |
|||
Do not forget to refresh the neighbor list whenever you change <math>\mathbf{H}\equiv [\mathbf{c}_1\, \mathbf{c}_2\, \mathbf{c}_3]</math> while atoms’ positions are fixed. MD++ automatically refreshes the neighbor list based on the change of the atoms’ positions. If atoms are fixed, the list will not be updated even if there could be a virtual displacement of atoms by changing <math>\mathbf{H}</math>. In the script (given at the end of this manual), you will see the command '''clearR0''', right after '''changeH_keepR''', which enforces the neighbor list to be refreshed. There are more MD++ commands to be explained: '''saveH''' stores the current 3×3 matrix '''H''' to '''H0''' and '''restoreH''' restores '''H''' from '''H0'''. '''RHtoS''' converts the real coordinate <math>\mathbf{r}_i</math> of an atom <math>i</math> to the scaled coordinate <math>\mathbf{s}_i</math> by <math>\mathbf{s}_i = \mathbf{H}^{-1} \mathbf{r}_i</math>. |
|||
|<math>\mathbf{c}_i := \mathbf{c}_i + frac \cdot \mathbf{c}_j </math>. |
|||
|} |
|||
Since we want to change the length of <math>\mathbf{c}_1</math> without changing its direction, both ''i'' and ''j'' should be 1 for computing the ideal tensile strength along <math>x</math> direction. (''i'' and ''j'' will be different when computing the ideal shear strength as described in the next section.) A related command '''changeH_keepS''' performs identical transformations to the repeat vectors, but the scaled coordinates, instead of the real coordinates of the atoms are kept fixed. This will lead to an affine transformation of the simulation cell and is related to the first definition of the ideal strength as described above. |
|||
After the '''changeH_keepR''', the potential energy is computed by the command '''eval'''. However, before computing the potential energy, we need to refresh the neighbor list. MD++ uses the Verlet list algorithm, in which the neighbor list is automatically refreshed whenever any atom is displaced by more than half the skin of the Verlet list since the last time the neighbor list is constructed. However, since here the atom positions do not change when we change the repeat vectors, the neighbor list will not be refreshed automatically. We can force the neighbor list to refresh by calling '''clearR0''' right after the '''changeH_keepR''' command. |
|||
The MD++ script for this calculation is attached at the end of this manual. A few more commands are used there to facilitate the calculation. The '''saveH''' command saves the current value of the <math>\mathbf{H}</math> matrix to <math>\mathbf{H}_0</math>. The '''restoreH''' command copies <math>\mathbf{H}_0</math> back to <math>\mathbf{H}</math>. This commands are used to make the matrix <math>\mathbf{H}</math> is always restored to the original value whenever the '''changeH_keepR''' command is called. The '''RHtoS''' command is always called before '''eval''' which converts the real coordinates <math>\mathbf{r}_i</math> of the atoms to the scaled coordinates <math>\mathbf{s}_i</math>, <math>\mathbf{s}_i = \mathbf{H}^{-1} \mathbf{r}_i</math>. This is needed because the calculation of potential energy in MD++ is based on the scaled coordinates. |
|||
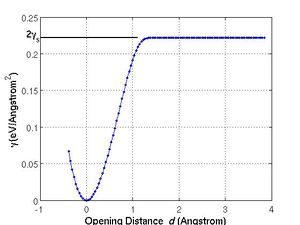
If we plot <math>\gamma</math> vs. <math>d</math>, the curve looks like Fig. 2(a). As <math>d</math> increases, the energy also increases up to a plateau <math>2\gamma_s</math>, where <math>\gamma_s</math> is the (unrelaxed) surface energy of (1 1 0) plane of Si. Once the opening distance becomes longer than certain cutoff distance, the interaction between two parts no longer plays a role and the energy becomes constant and irrelative to <math>d</math>. You may be aware that the slope of the <math>\gamma(d)</math> curve has the unit of stress. We define the maximum value of the slope as the ideal tensile strength along the separation direction, ''e.g.'' [1 1 0]. The SW potential model gives <math>\sigma_{[110]}^{\mathrm{th}} </math>=41.7 (GPa) for Si. |
|||
<gallery caption="Fig.1" widths="300px" heights="300px" perrow="2"> |
<gallery caption="Fig.1" widths="300px" heights="300px" perrow="2"> |
||
Image:Cut-surfL.jpg|(a) |
Image:Cut-surfL.jpg|(a) Creating a gap between the primary and image crystals on a (1 1 0) plane by changing the length of repeat vector <math>\mathbf{c}_1</math>. |
||
Image:Cut-slip.jpg|(b) |
Image:Cut-slip.jpg|(b) Sliding the primary and image crystals w.r.t. each other on a (1 1 1) plane by tilting the repeat vector <math>\mathbf{c}_2</math> along the <math>\mathbf{c}_1</math> direction.<ref name="kangcai2007"/> |
||
</gallery> |
|||
Fig. 2(a) plots the excess energy per unit area of the simulation cell, <math>\gamma</math>, as a function of separation distance <math>d</math> for the Stillinger-Weber (SW) model of Si. For large enough <math>d</math>, <math>\gamma(d)</math> reaches a plateau, whose value is <math>2\gamma_s</math>, where <math>\gamma_s</math> is the (unrelaxed) surface energy of (1 1 0) plane of Si. The plateau is reached when <math>d</math> becomes greater than the cut-off distance of the interatomic potential. The slope of the <math>\gamma(d)</math> has the unit of stress. The maximum slope of this curve can be interpreted as the maximum stress required to separate two rigid blocks of Si along the [1 1 0] direction, i.e. the ideal tensile strength. For the SW model of Si, <math>\sigma_{[110]}^{\mathrm{th}}</math> = 41.7 GPa. The same procedure described here can also be applied to compute ideal tensile strength by first-principles methods such as VASP. The shell script for this calculation is attached at the end of this manual. |
|||
<gallery caption="Fig.2" widths="300px" heights="300px" perrow="2"> |
|||
Image:Sisw_surfE.jpg|(a) The excess energy per unit area as a function of the opening distance for the SW model of Si. |
|||
Image:Sisw_slipE.jpg|(b) The excess energy per unit area as a function of the slip distance for the SW model of Si. |
|||
</gallery> |
</gallery> |
||
== Ideal Shear Strength == |
== Ideal Shear Strength == |
||
The procedure of calculating ideal shear strength is similar to that of ideal tensile strength except that we usually need to deal with two degrees of freedom. One is the amount of slip <math>d</math> in the plane of interest, and the other is the amount of openining <math>\delta</math> in the direction normal to the plane. The reason is that we usually need to allow <math>\delta</math> to relax when computing the ideal shear strength. |
|||
The procedure of calculating ideal shear strength is pretty much similar to that for |
|||
ideal tensile strength except that we now have to deal with 2-dimensional array |
|||
In this example, we will compute the ideal shear strength on a (1 1 1) plane of Si along the <math>[1 0 \bar{1}]</math> direction, <math>\sigma^{\mathrm{th}}_{(111)[10\bar{1}]}</math>. We start by creating a perfect crystal with repeat vectors |
|||
of energy. In Fig. 1(b), it is illustrated how we slip part of a bulk crystal to the |
|||
<math>\mathbf{c}_1 = N_x[1 0 \bar{1}]</math>, <math>\mathbf{c}_2 = N_y[1 1 1]</math> and <math>\mathbf{c}_3 = N_z[1 \bar{2} 1]</math>. |
|||
of all the atoms fixed, redefining <math>\mathbf{c}_2</math> vector as |
|||
Next, we perform the following change to the repeat vector <math>\mathbf{c}_2</math> while keeping the real coordinates of the atoms fixed, |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 70: | Line 78: | ||
|} |
|} |
||
The excess energy per unit area is now a function of both the slip distance <math>d</math> and the openning distance <math>\delta</math> |
|||
makes <math>\mathbf{c}_2</math> vector tilted and as a result the periodic image (2) will be shifted and moved from the crystal (1) in the primary cell by <math>d</math> and <math>\delta</math>, respectively. Notice that we do not simply slip but also allow the crystal move normal to slip plane and the energy <math>\gamma</math> is a function of <math>d</math> and <math>\delta</math> or |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
| Line 76: | Line 84: | ||
|} |
|} |
||
For a given <math>d</math>, we can let the opening distance <math>\delta</math> to relax, which leads to a one dimensional function |
|||
{|border="0" align="center" |
{|border="0" align="center" |
||
|<math> \Gamma = \displaystyle\min_{\{\delta\} }\gamma ( d ) </math>. |
|<math> \Gamma = \displaystyle\min_{\{\delta\} }\gamma ( d ) </math>. |
||
|} |
|} |
||
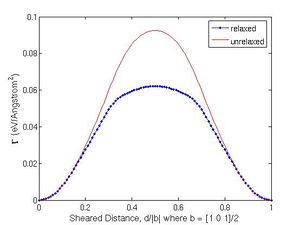
The minimization is performed numerically outside MD++, usually within Matlab, based on the 2-dimensional array output from MD++. Fig. 2(b) plots the function <math>\Gamma(d)</math> for SW model of Si. Due to the periodicity of the lattice, <math>\Gamma(d)</math> is a periodic function, with the periodicity being the length of the Burgers vector. Again, the slope of the <math>\Gamma(d)</math> curve also has the unit of stress. The maximum slope is defined as the ideal shear strength, which is 9.5 GPa for the SW model of Si. |
|||
The curve <math>\Gamma(d)</math> is shown in Fig. 2(b). The shifted distance <math>d</math> is normalized by the magnitude of Burgers vector and we observe that the misfit energy is |
|||
periodic after one Burgers vector. The ideal shear strength is also defined as the |
|||
maximum slope of the misfit energy curve and it is 9.5 GPa from SW potential model. |
|||
There are two different sets of (1 1 1) planes in a diamond-cubic crystal: the shuffle-set and the glide-set<ref name="kangcai2007"/>. Since the sliding is localized between the atomic planes at the edge of the simulation cell, whether or not the ideal shear strength is computed on the shuffle-set or the glide-set plane depends on which plane is exposed at the edge of the simulation cell. This can be changed by periodically shifting the atomic structure within the simulation cell (through command '''pbcshiftatom''') before calling '''changeH_keepR'''. The syntax for command '''pbcshfiftatom''' is |
|||
You have to keep it in mind that the shuffle set plane of (1 1 1) is your slip |
|||
plane. Fig. 3 shows two configurations which would be same unless you tilt |
|||
the periodicity vector. After the vector <math>\mathbf{c}_2</math> is tilted, one gives |
|||
<math>\Gamma</math> of shuffle-set plane and the other gives that of glide-set plane. And the ideal shear strength of glide-set plane (''e.g.'' 77.3GPa from SW) is much higher than that of shuffle-set plane. In MD++ script, this is handled by the command '''pbcshiftatom''', which shifts whole atoms periodically by '''dx''', '''dy''', and '''dz''' in all three directions. |
|||
input = [dx dy dz] pbcshiftatom |
input = [ ''dx'' ''dy'' ''dz'' ] pbcshiftatom |
||
where ''dx'', ''dy'' and ''dz'' are scaled coordinates of the shift vector. Since the [1 1 1] direction is along the <math>\mathbf{c}_2</math> direction, ''dy'' is the relevant parameter that will change the type of (1 1 1) plane exposed at the edge of the simulation cell. Whether or not '''pbcshiftatom''' is required and, if so, the necessary magnitude of ''dy'' can be determined by visually inspect the atomic structure in the X-window. The curves in Fig. 3(a) correspond to the shuffle-set plane<ref name="kangcai2007"/>. The ideal shear strength on the glide-set plane (77.3 GPa for SW model of Si) is much higher than that on the shuffle-set plane. |
|||
The numbers '''dx''', '''dy''' and '''dz''' are given in scaled coordinate. |
|||
<gallery caption="Fig.3" widths="300px" heights="300px" perrow="2"> |
|||
In MD++, there is a command '''calmisfit''' calculating the misfit energy <math>\gamma</math> as <math>d</math> and <math>\delta</math> change. |
|||
Image:Glideset_e.jpg |(a) A diamond-cubic structure with glide-set (1 1 1) plane exposed at the edge of the simulation cell (normal to <math>\mathbf{c}_2</math>. |
|||
Image:Shuffleset.jpg |(b) A diamond-cubic structure with shuffle-set (1 1 1) plane exposed at the edge of the simulation cell. The structures in (a) and (b) are identical unless slip occurs at the edge of the simulation cell by tilting repeat vector <math>\mathbf{c}_2</math>. |
|||
</gallery> |
|||
== Gamma Surface == |
|||
The above procedure allows us to compute the ideal shear strength on a specified plane along a specific slip direction. However, there are two orthogonal directions within every plane along which slip can occur. The excess energy per unit area as a function of the two-dimensional slip vector can be visualized as a surface, and is usually called the <math>\gamma</math>-surface, or the generalized stacking fault energy. |
|||
We have implemented a command '''calmisfit''' in MD++ that, together with an external Matlab program, computes the <math>\gamma</math>-surface. The command '''calmisfit''' itself computes the misfit energy <math>\gamma</math> as a function of three parameters: two for the 2-dimensional slip vector within the plane and one for the opening displacement <math>\delta</math> perpendicular to the plane. The minimization w.r.t. <math>\delta</math> is performed in the Matlab program. The syntax for this command is |
|||
<pre> |
<pre> |
||
| Line 105: | Line 115: | ||
</pre> |
</pre> |
||
The input variable needs |
The input variable needs ten elements, the first of which indicates the surface normal index. For example, if ''surfacenormal'' = 2, the slip plane is normal to the <math>\mathbf{c}_2</math> vector. The following nine numbers specifies the range and incremental steps of the offset vector in three directions. In the case study shown in Figs. 1(b) and 2(b), ''x0'', ''dx'', ''x1'' specify the range of <math>d</math>, in units of <math>|\mathbf{c}_1|/N_x</math>. ''y0'', ''dy'', ''y1'' specify the range of <math>\delta</math>, in units of <math>|\mathbf{c}_2|/N_y</math>. Since we are not interested in slip along the <math>\mathbf{c}_3</math> direction, we have set '''z0''' = '''dz''' = '''z1''' = 0. |
||
direction and how much the periodicity vector is tilted (or stretched). The numbers are normalized by the length of one lattice vector in each direction. If '''dx'''=0.001, the increment of tilting the vector in <math>x</math>-direction would be <math>0.001 \times a \times |\mathbf{c}_1|/N_x</math>, where <math>a</math> is the lattice constant. If '''dy'''=0.01, the vector <math>\mathbf{c}_2</math> increases by the amount of <math>0.01 \times a \times |\mathbf{c}_2|/N_y</math> in <math>y</math> direction. In our example, the last |
|||
three numbers are all zeros or '''z0''' = '''dz''' = '''z1''' = 0. |
|||
Command '''calmisfit''' produces a text output file “Emisfit.out”, which is then analyzed by the Matlab program cal_idealstr.m to perform minimization w.r.t. <math>\delta</math> and to compute the ideal shear strength. The source code for the cal_idealstr.m program is attached at the end of this manual. |
|||
After running '''calmisfit''', a text file “Emisfit.out” will be dumped out and it |
|||
will be used to calculate the ideal shear strength by the Matlab script attached |
|||
at the end. |
|||
== Ideal Shear Strength |
== Computing Ideal Shear Strength without Tilting Simulation Cell == |
||
For technical reasons, sometimes we cannot tilt the repeat vectors in MD++ when computing the ideal shear strength. For example, the ''meam-baskes'' program requires linking the C-programs of MD++ with a set of Fortran programs (provided by Dr. Mike Baskes) that implements the MEAM potential. Unfortunately, the three repeat vectors are not allowed to tilt arbitrarily in these Fortran programs. In this case, the approach described above will no longer apply. A new algorithm is needed to compute the ideal shear strength and the <math>\gamma</math>-surface. This is implemented in command '''calmisfit2'''. |
|||
Sometimes we simply cannot tilt the periodicity vectors. For example, the |
|||
original Baskes’ MEAM codes are implemented only for a rectangular cell. In |
|||
this case, the procedures explained above can not be applied and we have to |
|||
move a chunk of atoms literally. Fig. 4(a) shows the initial configuration in |
|||
which a few layers at top and bottom are removed in order to have room for |
|||
atoms to be shifted as shown in Fig. 4(b). Other than that, the rest of process |
|||
to get the ideal shear strength is identical. |
|||
'''calmisfit2''' litterally moves one block of atoms w.r.t. to the remaining block of atoms, as shown in Fig. 4(a). This creates an interface between the two blocks of atoms. To avoid creating another (unwanted) interface at the periodic boundary, we need to remove several layers of atoms at the edge of the simulation cell before calling '''calmisfit2'''. |
|||
To remove top and bottom atoms, two MD++ commands '''fixatoms_by_position''' |
|||
and '''removefixedatoms''' are used. The command '''fixatoms_by_position''' fixes |
|||
atoms within a hexahedral volume defined by six numbers ('''x0''', '''x1''', '''y0''', '''y1''', '''z0''' and '''z1''' in scaled coordinate) in input |
|||
Fig. 4(a) shows the initial configuration in which a few layers at top and bottom are removed in order to have room for atoms to be shifted as shown in Fig. 4(b). Other than that, the rest of process to get the ideal shear strength is identical. |
|||
input = [enable x0 x1 y0 y1 z0 z1 ] fixatoms_by_position |
|||
In MD++, we can remove a block of atom using commands '''fixatoms_by_position''' and '''removefixedatoms'''. The syntax is |
|||
atoms. |
|||
input = [''enable x0 x1 y0 y1 z0 z1'' ] fixatoms_by_position removefixedatoms |
|||
MD++ codes have another command '''calmisfit2''' to calculate the misfit energy |
|||
in non-tilting cell. The input variable for '''calmisfit2''' has six more numbers |
|||
''enable'' = 1 is required to activate the command, and the following 6 parameters specifies a parallelepiped volume (in scaled coordinates). All atoms within this volume will be labeled as fixed by the command '''fixatoms_by_positions'''. The following command '''removefixedatoms''' then removes these atoms. To remove two blocks of atoms at the top and bottom of the simulation cell, as shown in Fig. 4(a), the above commands need to be called twice with different input parameters. |
|||
('''xmin''', '''xmax''', '''ymin''', '''ymax''', '''zmin''' and '''zmax''') than '''calmisfit''' to define a hexahedral volume which will be moved by the command. Those numbers should be given in scaled coordinate. |
|||
The syntax for calling command '''calmisfit2''' is the following, |
|||
<pre> |
<pre> |
||
| Line 144: | Line 143: | ||
</pre> |
</pre> |
||
The first ten parameters have the same meaning as those for '''calmisfit'''. The last 6 parameters specify the parallelepiped volume (enclosed by the solid lines in Fig. 4(a)) which will be shifted w.r.t. the remaining part of the crystal. The command '''calmisfit2''' also produces a text file, “Emisfit.out”, which has the same format as that produced by '''calmisfit'''. The same Matlab program (cal_idealstr.m) is able to compute the ideal strength using it as the input. |
|||
The command '''calmisfit2''' also produces a text file, “Emisfit.out” of the same |
|||
format and the same Matlab script will read it to calculate the ideal shear |
|||
You may try '''calmisfit2_rigidrlx'''. Its syntax is given below. |
|||
strength. |
|||
<pre> |
|||
input = [ surfacenormal |
|||
x0 dx x1 |
|||
y0 dy y1 |
|||
z0 dz z1 |
|||
xmin xmax ymin ymax zmin zmax |
|||
nogrp grpid] |
|||
calmisfit2_rigidrlx |
|||
</pre> |
|||
Here, two more input parameters are added. The atoms in xmin<math>\le</math>sx<xmax, ymin<math>\le</math>sy<ymax, and zmin<math>\le</math>sz<zmax are grouped and their group ID is given as '''grpid'''. And the number of groups '''nogrp''' = 1 in most cases, here. The command '''calmisfit2_rigidrlx''' rigidly relaxes the group of atoms labeled as '''grpid''' along the surface normal direction. So, the "Emisfit.out" already contains relaxed surface energy. |
|||
<gallery caption="Fig.4" widths="300px" heights="300px" perrow="2"> |
|||
Image:Beforeshift_e.jpg |(a) Atomistic structure for computing ideal shear strength when the periodic repeat vectors are not allowed to tilt. Several layers of atoms at the top and bottom of the simulation cell are removed. The atoms inside the volume enclosed by the solid lines will be displaced w.r.t. the remaining atoms. |
|||
Image:Aftershift.jpg |(b) Atomistic structure after the atoms inside the volume specified in (a) are displaced. |
|||
</gallery> |
|||
== Script == |
== Script == |
||
The MD++ script and the Matlab |
The MD++ script (idealstr.tcl) and the Matlab program (cal_idealstr.m) are attached below. You can use them to reproduce the results described above. For example, |
||
to reproduce the results or try to calculate the ideal strength of other crystal |
|||
materials. Let’s say MD++ script file name is “idealstr.tcl”. If you type |
|||
$ bin/sw_gpp scripts/work/idealstr.tcl 0 |
$ bin/sw_gpp scripts/work/idealstr.tcl 0 |
||
computes the excess energy per unit area as a function of openning displacement perpendicular to the (1 1 0) plane. On the other hand, |
|||
<math>d</math>. If you type |
|||
$ bin/sw_gpp scripts/work/idealstr.tcl 1 |
$ bin/sw_gpp scripts/work/idealstr.tcl 1 |
||
computes the misfit energy of (1 1 1) plane along <math>[1\, 0\, \bar{1}]</math> direction by tilting the periodic repeat vector <math>\mathbf{c}_2</math>. Finally, |
|||
$ bin/sw_gpp scripts/work/idealstr.tcl 2 |
$ bin/sw_gpp scripts/work/idealstr.tcl 2 |
||
computes the same misfit energy as above without tilting the periodic repeat vector. |
|||
After performing the above MD++ calculation, several files will be generated in the output directory, including Esurf.dat (for tensile strength) and Emisfit.dat (for shear strength). Copying the Matlab program into the same directory and running this Matlab program will produce energy curves and ideal strength values reported as above. |
|||
=== MD++ Script === |
|||
=== MD++ Input Script idealstr.tcl === |
|||
<pre> |
<pre> |
||
# -*-shell-script-*- |
# -*-shell-script-*- |
||
| Line 319: | Line 334: | ||
openwindow |
openwindow |
||
MD++ sleep quit |
MD++ sleep quit |
||
MD++ quit |
|||
} elseif { $status == 3 } { |
|||
# calculate generalized stacking fault energy on (1 1 1) along [1 0 -1] w/ calmisfit2_rigidrlx |
|||
initmd |
|||
# surface normal direction 1/x 2/y 3/z |
|||
set surfacenormal 2 |
|||
set lattconst 5.431 |
|||
set c1 "1 0 -1"; set c2 "1 1 1"; set c3 "1 -2 1" |
|||
set Nx 5; set Ny 6; set Nz 3 |
|||
set gridx " 0.000 0.005 0.500" |
|||
set gridy " 0.000 0.010 0.000" |
|||
set gridz " 0.000 0.050 0.000" |
|||
set xmin -1.00; set xmax 1.00 |
|||
set ymin 0.01; set ymax 1.00 |
|||
set zmin -1.00; set zmax 1.00 |
|||
set grp_id 5 |
|||
# write the matlab script |
|||
set fileID [open "parameter.m" w] |
|||
puts $fileID "normaldir = $surfacenormal" |
|||
puts $fileID "latticeconst = $lattconst" |
|||
puts $fileID "latticesize = \[ $c1 $Nx " |
|||
puts $fileID " $c2 $Ny " |
|||
puts $fileID " $c3 $Nz \]" |
|||
puts $fileID "dxyz = \[ $gridx " |
|||
puts $fileID " $gridy " |
|||
puts $fileID " $gridz \]" |
|||
close $fileID |
|||
MD++ { |
|||
element0 = Si |
|||
crystalstructure = diamond-cubic |
|||
} |
|||
MD++ latticeconst = $lattconst |
|||
MD++ latticesize = \[$c1 $Nx $c2 $Ny $c3 $Nz\] |
|||
MD++ makecrystal |
|||
MD++ input = \[1 -1 1 0.4 1 -1 1 \] fixatoms_by_position removefixedatoms |
|||
MD++ input = \[1 -1 1 -1 -0.42 -1 1 \] fixatoms_by_position removefixedatoms |
|||
MD++ input = \[ $surfacenormal $gridx $gridy $gridz $xmin $xmax $ymin $ymax $zmin $zmax 1 $grp_id \] |
|||
MD++ calmisfit2_rigidrlx |
|||
MD++ quit |
MD++ quit |
||
| Line 327: | Line 386: | ||
</pre> |
</pre> |
||
=== Matlab |
=== Matlab Program cal_idealstr.m === |
||
After running MD++, you just run the following matlab script in the directory |
|||
where the output files exist. Then the matlab script will plot the energy curve |
|||
and calculate ideal strengths. |
|||
<pre> |
<pre> |
||
| Line 385: | Line 441: | ||
for i = 1:MM(1), |
for i = 1:MM(1), |
||
a = E(i,:); |
a = E(i,:); |
||
if length(a)> 1 |
|||
a_interp = interp1(normalgrid,a,normalfinegrid,’spline’); |
|||
a_interp = interp1(normalgrid,a,normalfinegrid,'spline'); |
|||
else |
|||
a_interp = a; |
|||
end |
|||
[Erlx(i), ind] = min(a_interp); |
[Erlx(i), ind] = min(a_interp); |
||
Zrlx(i) = normalfinegrid(ind); |
Zrlx(i) = normalfinegrid(ind); |
||
| Line 411: | Line 471: | ||
ylabel({’Lowest energy height,’; ’\delta/|c_2| where c_2 = [1 1 1]’},’fontsize’,16) |
ylabel({’Lowest energy height,’; ’\delta/|c_2| where c_2 = [1 1 1]’},’fontsize’,16) |
||
end |
end |
||
</pre> |
|||
=== Shell script for Computing Ideal Tensile strength by VASP === |
|||
The following shell script will create the '''POSCAR''' file which specifies the atomic positions for VASP. You will also need to prepare '''INCAR''', '''KPOINTS''', '''POTCAR''' files in the same directory for VASP to run. This script will run VASP in serial mode. You can modify it to submit VASP jobs in parallel. |
|||
In this script, the length of the first lattice vector is changed from 0.9 to 1.6 of its equilibrium length in steps of 0.01. A VASP calculation is performed for each length of the lattice vector to obtain the potential energy, which is then saved into file ''summary''. |
|||
<pre> |
|||
#!/bin/bash |
|||
declare -i x=9000 |
|||
while (($x <= 16000)) |
|||
do |
|||
a=$(echo "scale=3; $x/10000" | bc -l) |
|||
cat > POSCAR << FIN |
|||
Si comment line |
|||
5.389 universal scaling factor |
|||
$a $a 0.0 first lattice vector |
|||
-1.0 1.0 0.0 second lattice vector |
|||
0.0 0.0 1.0 third lattice vector |
|||
16 number of atoms per species |
|||
selective dynamics |
|||
cart direct or cart (only first letter is significant) |
|||
-0.5000000000000000 0.5000000000000000 0.0000000000000000 F F F |
|||
-0.2500000000000000 0.7500000000000000 0.2500000000000000 F F F |
|||
-0.7500000000000000 0.7500000000000000 0.7500000000000000 F F F |
|||
-0.2500000000000000 0.2500000000000000 0.7500000000000000 F F F |
|||
-0.5000000000000000 1.0000000000000000 0.5000000000000000 F F F |
|||
-0.2500000000000000 1.2500000000000000 0.7500000000000000 F F F |
|||
0.0000000000000000 0.0000000000000000 0.0000000000000000 F F F |
|||
0.5000000000000000 0.5000000000000000 0.0000000000000000 F F F |
|||
0.0000000000000000 0.5000000000000000 0.5000000000000000 F F F |
|||
0.2500000000000000 0.2500000000000000 0.2500000000000000 F F F |
|||
0.7500000000000000 0.7500000000000000 0.2500000000000000 F F F |
|||
0.2500000000000000 0.7500000000000000 0.7500000000000000 F F F |
|||
0.0000000000000000 1.0000000000000000 0.0000000000000000 F F F |
|||
0.0000000000000000 1.5000000000000000 0.5000000000000000 F F F |
|||
0.5000000000000000 1.0000000000000000 0.5000000000000000 F F F |
|||
0.2500000000000000 1.2500000000000000 0.2500000000000000 F F F |
|||
FIN |
|||
vasp |
|||
E=`tail -1 OSZICAR` |
|||
echo $a $E >> summary |
|||
let x+=100 |
|||
done |
|||
</pre> |
|||
=== Shell script for Computing Ideal Shear strength by VASP === |
|||
The following shell script will create the '''POSCAR''' file which specifies the atomic positions for VASP. You will also need to prepare '''INCAR''', '''KPOINTS''', '''POTCAR''' files in the same directory for VASP to run. This script will run VASP in serial mode. You can modify it to submit VASP jobs in parallel. |
|||
In this script, position of a set of atoms is displaced along the y-direction [1 0 -1] but are allowed to relax in the x-direction [1 1 1]. A VASP calculation is performed for each length of the lattice vector to obtain the potential energy, which is then saved into file ''summary''. |
|||
<pre> |
|||
#!/bin/bash |
|||
function add() { |
|||
echo "scale=17; $1 + $2" | bc -l |
|||
} |
|||
function subtract() { |
|||
echo "scale=17; $1 - $2" | bc -l |
|||
} |
|||
function div() { |
|||
echo "scale=3; $1 / $2" | bc -l |
|||
} |
|||
declare -i x=0 |
|||
while (($x <= 50)) |
|||
do |
|||
a=$(div $x 100) |
|||
cat > POSCAR << FIN |
|||
Si comment line |
|||
5.389 universal scaling factor |
|||
2.0 2.0 2.0 first lattice vector |
|||
1.0 0.0 -1.0 second lattice vector |
|||
-0.5 1.0 -0.5 third lattice vector |
|||
32 number of atoms per species |
|||
selective dynamics |
|||
direct direct or cart (only first letter is significant) |
|||
-0.3125000000000000 0.2500000000000000 0.0000000000000000 T F F |
|||
-0.3125000000000000 -0.2500000000000000 0.0000000000000000 T F F |
|||
-0.3125000000000000 0.0000000000000000 -0.5000000000000000 T F F |
|||
-0.2708333333333334 -0.2500000000000000 -0.3333333333333334 T F F |
|||
-0.3125000000000000 -0.5000000000000000 -0.5000000000000000 T F F |
|||
-0.1458333333333334 -0.2500000000000000 -0.3333333333333334 T F F |
|||
-0.2708333333333334 0.0000000000000000 0.1666666666666666 T F F |
|||
-0.1458333333333334 0.0000000000000000 0.1666666666666666 T F F |
|||
-0.2708333333333334 -0.5000000000000000 0.1666666666666666 T F F |
|||
-0.1041666666666667 -0.2500000000000000 0.3333333333333333 T F F |
|||
-0.1041666666666667 -0.5000000000000000 -0.1666666666666667 T F F |
|||
-0.1458333333333334 -0.5000000000000000 0.1666666666666666 T F F |
|||
0.0208333333333333 $(add -0.2500000000000000 $a) 0.3333333333333333 T F F |
|||
0.0208333333333333 $(add -0.5000000000000000 $a) -0.1666666666666667 T F F |
|||
-0.2708333333333334 0.2500000000000000 -0.3333333333333334 T F F |
|||
-0.1458333333333334 0.2500000000000000 -0.3333333333333334 T F F |
|||
-0.1041666666666667 0.2500000000000000 0.3333333333333333 T F F |
|||
0.0208333333333333 $(add 0.2500000000000000 $a) 0.3333333333333333 T F F |
|||
-0.1041666666666667 0.0000000000000000 -0.1666666666666667 T F F |
|||
0.0208333333333333 $(add 0.0000000000000000 $a) -0.1666666666666667 T F F |
|||
0.0625000000000000 $(add 0.2500000000000000 $a) 0.0000000000000000 T F F |
|||
0.0625000000000000 $(add -0.2500000000000000 $a) 0.0000000000000000 T F F |
|||
0.0625000000000000 $(add 0.0000000000000000 $a) -0.5000000000000000 T F F |
|||
0.1875000000000000 $(add 0.2500000000000000 $a) 0.0000000000000000 T F F |
|||
0.1875000000000000 $(add -0.2500000000000000 $a) 0.0000000000000000 T F F |
|||
0.1875000000000000 $(add 0.0000000000000000 $a) -0.5000000000000000 T F F |
|||
0.0625000000000000 $(add -0.5000000000000000 $a) -0.5000000000000000 T F F |
|||
0.2291666666666666 $(add -0.2500000000000000 $a) -0.3333333333333334 T F F |
|||
0.1875000000000000 $(add -0.5000000000000000 $a) -0.5000000000000000 T F F |
|||
0.2291666666666666 $(add 0.0000000000000000 $a) 0.1666666666666666 T F F |
|||
0.2291666666666666 $(add -0.5000000000000000 $a) 0.1666666666666666 T F F |
|||
0.2291666666666666 $(add 0.2500000000000000 $a) -0.3333333333333334 T F F |
|||
FIN |
|||
vasp |
|||
E=`tail -1 OSZICAR` |
|||
echo $a $E >> summary |
|||
let x+=5 |
|||
done |
|||
</pre> |
</pre> |
||
Latest revision as of 11:49, 21 November 2014
Manual 09 for MD++
Computing Ideal Strength
Keonwook Kang and Wei Cai
The ideal strength of a material is the maximum stress that it can sustain and is an upper bound to the critical stress for crack and dislocation nucleation in the material. When the applied stress exceeds the ideal strength, the crystal structure will collapse even at zero temperature. The mode of failure depends on the type of applied stress. When the tensile stress exceeds the ideal tensile strength, the crystal is unstable against cleavage fracture. When the shear stress exceeds the ideal shear strength, the crysal is unstable against slip on crystallographic planes. Because a real crystal is not perfect but contains defects (such as surfaces, grain boundaries, dislocations, vacancies), its strength is lower than the ideal strength. When the defect density is made sufficiently small (such as in whiskers), the strength of a real crystal can approach the ideal strength. The ideal strength is also called the theoretical strength.
There are two alternative definitions of the ideal strength depending on the way that the deformation is imposed on the perfect crystal structure. In the first definition, the deformation strain can be applied uniformly to the simulation cell (usually under periodic boundary conditions) resulting in an affine deformation of the entire crystal[1]. In the second definition, the deformation strain can be localized between two crystallographic planes[2]. The ideal strength values obtained from these two approaches are somewhat different but are also expected to co-relate with each other. In this manual, we describe how to compute the ideal strength according to the second definition, using diamond-cubic structure of Si as an example.
Ideal Tensile Strength
Suppose we want to compute the ideal tensile strength of Si along the [1 1 0] direction, i.e., . First, we create a simulation cell under periodic boundary condition (PBC) with the three periodicity vectors , and . The coordinate system is defined such that , and are aligned along Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle x} , Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle y} and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle z} directions, respectively. The crystal just created is surrounded by its own images in all three directions.
![{\displaystyle \sigma _{[110]}^{\mathrm {th} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0f75b0edd2d439abde70faadfaf47c6ffffca13)
![{\displaystyle \mathbf {c} _{1}=N_{x}[110]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fba118bbcc340e67c33f8ffc8dfc4826a4b8f581)
![{\displaystyle \mathbf {c} _{2}=N_{y}[{\bar {1}}10]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b435a061d19dc0fe0f28450cd5fa59df61447260)
![{\displaystyle \mathbf {c} _{3}=N_{z}[001]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a44758ea1bec16598a3cee23a5054cead46d07fd)



Next, we change the length of the repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1} while keeping the position of all atoms in the simulation cell fixed. Let Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1^0} be the original value of the repeat vector along the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle x} direction and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle L_x^0 = |\mathbf{c}_1^0|} . Suppose we assign the new repeat vector to be
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1 = \left( 1 + \frac{d}{L_x^0}\right) \mathbf{c}_1^0 } . |
This will create a gap of width Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} between the crystal in the simulation cell (primary cell) with its images in the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle x} direction, as shown in Fig. 1(a). For each value of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} , we compute the potential energy of the crystal (without relaxing the positions of the atoms). The excess energy with respect to the case of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d=0} , dividied by the cross section area of the simulation cell perpendicular to the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle x} direction, is defined as function of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d}
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma = \gamma ( d ) } . |
and is plotted in Fig. 2(a).
In MD++, the transformation of the simulation cell as described above can be performed conveniently using the command changeH_keepR. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}} is a 3 by 3 matrix whose three columns correspond to the coordinates of the three repeat vectors, i.e. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H} = [\mathbf{c}_1 | \mathbf{c}_2 | \mathbf{c}_3]} . The syntax for this command is
input = [ i j frac ] changeH_keepR
This will lead to the following transformation of the repeat vectors
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_i := \mathbf{c}_i + frac \cdot \mathbf{c}_j } . |
Since we want to change the length of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1} without changing its direction, both i and j should be 1 for computing the ideal tensile strength along Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle x} direction. (i and j will be different when computing the ideal shear strength as described in the next section.) A related command changeH_keepS performs identical transformations to the repeat vectors, but the scaled coordinates, instead of the real coordinates of the atoms are kept fixed. This will lead to an affine transformation of the simulation cell and is related to the first definition of the ideal strength as described above.
After the changeH_keepR, the potential energy is computed by the command eval. However, before computing the potential energy, we need to refresh the neighbor list. MD++ uses the Verlet list algorithm, in which the neighbor list is automatically refreshed whenever any atom is displaced by more than half the skin of the Verlet list since the last time the neighbor list is constructed. However, since here the atom positions do not change when we change the repeat vectors, the neighbor list will not be refreshed automatically. We can force the neighbor list to refresh by calling clearR0 right after the changeH_keepR command.
The MD++ script for this calculation is attached at the end of this manual. A few more commands are used there to facilitate the calculation. The saveH command saves the current value of the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}} matrix to Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}_0} . The restoreH command copies Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}_0} back to Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}} . This commands are used to make the matrix Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{H}} is always restored to the original value whenever the changeH_keepR command is called. The RHtoS command is always called before eval which converts the real coordinates Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{r}_i} of the atoms to the scaled coordinates Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{s}_i} , Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{s}_i = \mathbf{H}^{-1} \mathbf{r}_i} . This is needed because the calculation of potential energy in MD++ is based on the scaled coordinates.
- Fig.1
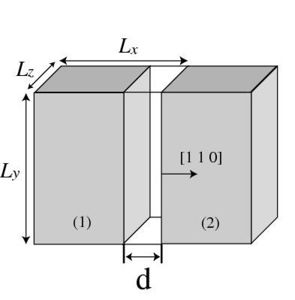
(a) Creating a gap between the primary and image crystals on a (1 1 0) plane by changing the length of repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1} .
![(b) Sliding the primary and image crystals w.r.t. each other on a (1 1 1) plane by tilting the repeat vector '"`UNIQ--postMath-00000025-QINU`"' along the '"`UNIQ--postMath-00000026-QINU`"' direction.[2]](/mediawiki/images/thumb/d/d2/Cut-slip.jpg/300px-Cut-slip.jpg)
(b) Sliding the primary and image crystals w.r.t. each other on a (1 1 1) plane by tilting the repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} along the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1} direction.[2]

![(b) Sliding the primary and image crystals w.r.t. each other on a (1 1 1) plane by tilting the repeat vector '"`UNIQ--postMath-00000025-QINU`"' along the '"`UNIQ--postMath-00000026-QINU`"' direction.[2]](/wiki/File:Cut-slip.jpg)
Fig. 2(a) plots the excess energy per unit area of the simulation cell, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma}
, as a function of separation distance Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d}
for the Stillinger-Weber (SW) model of Si. For large enough Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d}
, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma(d)}
reaches a plateau, whose value is Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle 2\gamma_s}
, where Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma_s}
is the (unrelaxed) surface energy of (1 1 0) plane of Si. The plateau is reached when Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d}
becomes greater than the cut-off distance of the interatomic potential. The slope of the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma(d)}
has the unit of stress. The maximum slope of this curve can be interpreted as the maximum stress required to separate two rigid blocks of Si along the [1 1 0] direction, i.e. the ideal tensile strength. For the SW model of Si, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma_{[110]}^{\mathrm{th}}}
= 41.7 GPa. The same procedure described here can also be applied to compute ideal tensile strength by first-principles methods such as VASP. The shell script for this calculation is attached at the end of this manual.
- Fig.2
(a) The excess energy per unit area as a function of the opening distance for the SW model of Si.
(b) The excess energy per unit area as a function of the slip distance for the SW model of Si.


Ideal Shear Strength
The procedure of calculating ideal shear strength is similar to that of ideal tensile strength except that we usually need to deal with two degrees of freedom. One is the amount of slip Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} in the plane of interest, and the other is the amount of openining Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} in the direction normal to the plane. The reason is that we usually need to allow Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} to relax when computing the ideal shear strength.
In this example, we will compute the ideal shear strength on a (1 1 1) plane of Si along the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [1 0 \bar{1}]} direction, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \sigma^{\mathrm{th}}_{(111)[10\bar{1}]}} . We start by creating a perfect crystal with repeat vectors Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_1 = N_x[1 0 \bar{1}]} , Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2 = N_y[1 1 1]} and Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_3 = N_z[1 \bar{2} 1]} .
Next, we perform the following change to the repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} while keeping the real coordinates of the atoms fixed,
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \begin{align} \mathbf{c}_2 &= \left(1+ \frac{\delta}{L_z^0}\mathbf{c}_2^0 + \frac{d}{L_x^0}\mathbf{c}_1^0 \right) \\ &= (L_z^0 + \delta)\mathbf{n} + d\mathbf{m} \end{align} } |
The excess energy per unit area is now a function of both the slip distance Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} and the openning distance Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta}
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma = \gamma ( d, \delta ) } . |
For a given Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} , we can let the opening distance Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} to relax, which leads to a one dimensional function
| Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Gamma = \displaystyle\min_{\{\delta\} }\gamma ( d ) } . |
The minimization is performed numerically outside MD++, usually within Matlab, based on the 2-dimensional array output from MD++. Fig. 2(b) plots the function Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Gamma(d)} for SW model of Si. Due to the periodicity of the lattice, Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Gamma(d)} is a periodic function, with the periodicity being the length of the Burgers vector. Again, the slope of the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Gamma(d)} curve also has the unit of stress. The maximum slope is defined as the ideal shear strength, which is 9.5 GPa for the SW model of Si.
There are two different sets of (1 1 1) planes in a diamond-cubic crystal: the shuffle-set and the glide-set[2]. Since the sliding is localized between the atomic planes at the edge of the simulation cell, whether or not the ideal shear strength is computed on the shuffle-set or the glide-set plane depends on which plane is exposed at the edge of the simulation cell. This can be changed by periodically shifting the atomic structure within the simulation cell (through command pbcshiftatom) before calling changeH_keepR. The syntax for command pbcshfiftatom is
input = [ dx dy dz ] pbcshiftatom
where dx, dy and dz are scaled coordinates of the shift vector. Since the [1 1 1] direction is along the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} direction, dy is the relevant parameter that will change the type of (1 1 1) plane exposed at the edge of the simulation cell. Whether or not pbcshiftatom is required and, if so, the necessary magnitude of dy can be determined by visually inspect the atomic structure in the X-window. The curves in Fig. 3(a) correspond to the shuffle-set plane[2]. The ideal shear strength on the glide-set plane (77.3 GPa for SW model of Si) is much higher than that on the shuffle-set plane.
- Fig.3
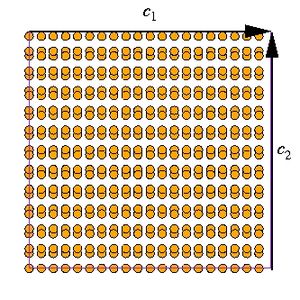
(a) A diamond-cubic structure with glide-set (1 1 1) plane exposed at the edge of the simulation cell (normal to Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} .

(b) A diamond-cubic structure with shuffle-set (1 1 1) plane exposed at the edge of the simulation cell. The structures in (a) and (b) are identical unless slip occurs at the edge of the simulation cell by tilting repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} .


Gamma Surface
The above procedure allows us to compute the ideal shear strength on a specified plane along a specific slip direction. However, there are two orthogonal directions within every plane along which slip can occur. The excess energy per unit area as a function of the two-dimensional slip vector can be visualized as a surface, and is usually called the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma} -surface, or the generalized stacking fault energy.
We have implemented a command calmisfit in MD++ that, together with an external Matlab program, computes the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma} -surface. The command calmisfit itself computes the misfit energy Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma} as a function of three parameters: two for the 2-dimensional slip vector within the plane and one for the opening displacement Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} perpendicular to the plane. The minimization w.r.t. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} is performed in the Matlab program. The syntax for this command is
input = [ surfacenormal
x0 dx x1
y0 dy y1
z0 dz z1 ]
calmisfit
The input variable needs ten elements, the first of which indicates the surface normal index. For example, if surfacenormal = 2, the slip plane is normal to the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} vector. The following nine numbers specifies the range and incremental steps of the offset vector in three directions. In the case study shown in Figs. 1(b) and 2(b), x0, dx, x1 specify the range of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle d} , in units of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle |\mathbf{c}_1|/N_x} . y0, dy, y1 specify the range of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} , in units of Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle |\mathbf{c}_2|/N_y} . Since we are not interested in slip along the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_3} direction, we have set z0 = dz = z1 = 0.
Command calmisfit produces a text output file “Emisfit.out”, which is then analyzed by the Matlab program cal_idealstr.m to perform minimization w.r.t. Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \delta} and to compute the ideal shear strength. The source code for the cal_idealstr.m program is attached at the end of this manual.
Computing Ideal Shear Strength without Tilting Simulation Cell
For technical reasons, sometimes we cannot tilt the repeat vectors in MD++ when computing the ideal shear strength. For example, the meam-baskes program requires linking the C-programs of MD++ with a set of Fortran programs (provided by Dr. Mike Baskes) that implements the MEAM potential. Unfortunately, the three repeat vectors are not allowed to tilt arbitrarily in these Fortran programs. In this case, the approach described above will no longer apply. A new algorithm is needed to compute the ideal shear strength and the Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \gamma} -surface. This is implemented in command calmisfit2.
calmisfit2 litterally moves one block of atoms w.r.t. to the remaining block of atoms, as shown in Fig. 4(a). This creates an interface between the two blocks of atoms. To avoid creating another (unwanted) interface at the periodic boundary, we need to remove several layers of atoms at the edge of the simulation cell before calling calmisfit2.
Fig. 4(a) shows the initial configuration in which a few layers at top and bottom are removed in order to have room for atoms to be shifted as shown in Fig. 4(b). Other than that, the rest of process to get the ideal shear strength is identical.
In MD++, we can remove a block of atom using commands fixatoms_by_position and removefixedatoms. The syntax is
input = [enable x0 x1 y0 y1 z0 z1 ] fixatoms_by_position removefixedatoms
enable = 1 is required to activate the command, and the following 6 parameters specifies a parallelepiped volume (in scaled coordinates). All atoms within this volume will be labeled as fixed by the command fixatoms_by_positions. The following command removefixedatoms then removes these atoms. To remove two blocks of atoms at the top and bottom of the simulation cell, as shown in Fig. 4(a), the above commands need to be called twice with different input parameters.
The syntax for calling command calmisfit2 is the following,
input = [ surfacenormal
x0 dx x1
y0 dy y1
z0 dz z1
xmin xmax ymin ymax zmin zmax]
calmisfit2
The first ten parameters have the same meaning as those for calmisfit. The last 6 parameters specify the parallelepiped volume (enclosed by the solid lines in Fig. 4(a)) which will be shifted w.r.t. the remaining part of the crystal. The command calmisfit2 also produces a text file, “Emisfit.out”, which has the same format as that produced by calmisfit. The same Matlab program (cal_idealstr.m) is able to compute the ideal strength using it as the input.
You may try calmisfit2_rigidrlx. Its syntax is given below.
input = [ surfacenormal
x0 dx x1
y0 dy y1
z0 dz z1
xmin xmax ymin ymax zmin zmax
nogrp grpid]
calmisfit2_rigidrlx
Here, two more input parameters are added. The atoms in xminFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \le} sx<xmax, yminFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \le} sy<ymax, and zminFailed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \le} sz<zmax are grouped and their group ID is given as grpid. And the number of groups nogrp = 1 in most cases, here. The command calmisfit2_rigidrlx rigidly relaxes the group of atoms labeled as grpid along the surface normal direction. So, the "Emisfit.out" already contains relaxed surface energy.
- Fig.4
(a) Atomistic structure for computing ideal shear strength when the periodic repeat vectors are not allowed to tilt. Several layers of atoms at the top and bottom of the simulation cell are removed. The atoms inside the volume enclosed by the solid lines will be displaced w.r.t. the remaining atoms.

(b) Atomistic structure after the atoms inside the volume specified in (a) are displaced.


Script
The MD++ script (idealstr.tcl) and the Matlab program (cal_idealstr.m) are attached below. You can use them to reproduce the results described above. For example,
$ bin/sw_gpp scripts/work/idealstr.tcl 0
computes the excess energy per unit area as a function of openning displacement perpendicular to the (1 1 0) plane. On the other hand,
$ bin/sw_gpp scripts/work/idealstr.tcl 1
computes the misfit energy of (1 1 1) plane along Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [1\, 0\, \bar{1}]} direction by tilting the periodic repeat vector Failed to parse (SVG (MathML can be enabled via browser plugin): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \mathbf{c}_2} . Finally,
$ bin/sw_gpp scripts/work/idealstr.tcl 2
computes the same misfit energy as above without tilting the periodic repeat vector.
After performing the above MD++ calculation, several files will be generated in the output directory, including Esurf.dat (for tensile strength) and Emisfit.dat (for shear strength). Copying the Matlab program into the same directory and running this Matlab program will produce energy curves and ideal strength values reported as above.
MD++ Input Script idealstr.tcl
# -*-shell-script-*-
source "scripts/Examples/Tcl/startup.tcl"
#*******************************************
# Definition of procedures
#*******************************************
proc initmd { } {
MD++ setnolog
MD++ setoverwrite
MD++ dirname = runs/si_idealstr
MD++ NIC = 200 NNM = 200
}
proc openwindow { } { MD++ {
# Plot Configuration
atomradius = 0.67 bondradius = 0.3 bondlength = 0
atomcolor = orange bondcolor = red backgroundcolor = gray70
win_width = 300 win_height = 300
plotfreq = 10 rotateangles = [ 0 0 0 1.25 ]
plot_atom_info = 1 # reduced coordinates of atoms
# plot_atom_info = 2 # real coordinates of atoms
# plot_atom_info = 3 # energy of atoms
openwin alloccolors rotate saverot eval plot
} }
#*******************************************
# Main program starts here
#*******************************************
if { $argc == 0 } {
set status 0
} elseif { $argc > 0 } {
set status [lindex $argv 0]
}
puts "status = $status"
if { $status == 0 } {
# calculate surface opening energy on (1 1 0).
initmd
MD++ {
element0 = Si latticeconst = 5.43095
crystalstructure = diamond-cubic
latticesize = [ 1 1 0 5
-1 1 0 5
0 0 1 7 ]
makecrystal }
MD++ saveH eval
set Lx0 [MD++_Get H_11]
set Ly0 [MD++_Get H_22]
set Lz0 [MD++_Get H_33]
set EPOT0 [MD++_Get EPOT]
set Area [expr $Ly0 * $Lz0]
set fileID [open "Esurf.dat" w]
for {set i -10} {$i <= 100} {incr i} {
set strain [expr $i/1000.0]
MD++ restoreH RHtoS
MD++ input = \[ 1 1 $strain \] changeH_keepR
MD++ clearR0 # enforce refresh neighbor list
MD++ eval
set Lx [MD++_Get H_11]
set dist [expr $Lx - $Lx0]
set EPOT [MD++_Get EPOT]
# Esurf2 : excessive energy per area to create 2 free surfaces.
set Esurf2 [expr ($EPOT - $EPOT0)/$Area]
puts $fileID "[format %18.14e $dist]\t \
[format %18.14e $Esurf2]"
}
close $fileID
MD++ quit
} elseif { $status == 1 } {
# calculate generalized stacking fault energy on (1 1 1) along [1 0 -1]
initmd
# surface normal direction 1/x 2/y 3/z
set surfacenormal 2
set lattconst 5.43095
set c1 "1 0 -1"; set c2 "1 1 1"; set c3 "1 -2 1"
set Nx 5; set Ny 4; set Nz 3
set gridx " 0.000 0.005 0.500"
set gridy "-0.050 0.010 0.150"
set gridz " 0.000 0.050 0.000"
# write the matlab script
set fileID [open "parameter.m" w]
puts $fileID "normaldir = $surfacenormal"
puts $fileID "latticeconst = $lattconst"
puts $fileID "latticesize = \[ $c1 $Nx "
puts $fileID " $c2 $Ny "
puts $fileID " $c3 $Nz \]"
puts $fileID "dxyz = \[ $gridx "
puts $fileID " $gridy "
puts $fileID " $gridz \]"
close $fileID
MD++ {
element0 = Si
crystalstructure = diamond-cubic
}
MD++ latticeconst = $lattconst
MD++ latticesize = \[$c1 $Nx $c2 $Ny $c3 $Nz\]
MD++ makecrystal
# To make shuffle set of (111) plane exposed
MD++ { input = [0 0.05 0 ] pbcshiftatom }
MD++ input = \[ $surfacenormal $gridx $gridy $gridz \]
MD++ calmisfit
MD++ quit
} elseif { $status == 2 } {
# calculate generalized stacking fault energy on (1 1 1) along [1 0 -1]
initmd
# surface normal direction 1/x 2/y 3/z
set surfacenormal 2
set lattconst 5.431
set c1 "1 0 -1"; set c2 "1 1 1"; set c3 "1 -2 1"
set Nx 5; set Ny 6; set Nz 3
set gridx " 0.000 0.005 0.500"
set gridy "-0.050 0.010 0.150"
set gridz " 0.000 0.050 0.000"
set xmin -1.00; set xmax 1.00
set ymin 0.01; set ymax 1.00
set zmin -1.00; set zmax 1.00
# write the matlab script
set fileID [open "parameter.m" w]
puts $fileID "normaldir = $surfacenormal"
puts $fileID "latticeconst = $lattconst"
puts $fileID "latticesize = \[ $c1 $Nx "
puts $fileID " $c2 $Ny "
puts $fileID " $c3 $Nz \]"
puts $fileID "dxyz = \[ $gridx "
puts $fileID " $gridy "
puts $fileID " $gridz \]"
close $fileID
MD++ {
element0 = Si
crystalstructure = diamond-cubic
}
MD++ latticeconst = $lattconst
MD++ latticesize = \[$c1 $Nx $c2 $Ny $c3 $Nz\]
MD++ makecrystal
MD++ input = \[1 -1 1 0.4 1 -1 1 \] fixatoms_by_position removefixedatoms
MD++ input = \[1 -1 1 -1 -0.42 -1 1 \] fixatoms_by_position removefixedatoms
MD++ input = \[ $surfacenormal $gridx $gridy $gridz $xmin $xmax $ymin $ymax $zmin $zmax \]
MD++ calmisfit2
openwindow
MD++ sleep quit
MD++ quit
} elseif { $status == 3 } {
# calculate generalized stacking fault energy on (1 1 1) along [1 0 -1] w/ calmisfit2_rigidrlx
initmd
# surface normal direction 1/x 2/y 3/z
set surfacenormal 2
set lattconst 5.431
set c1 "1 0 -1"; set c2 "1 1 1"; set c3 "1 -2 1"
set Nx 5; set Ny 6; set Nz 3
set gridx " 0.000 0.005 0.500"
set gridy " 0.000 0.010 0.000"
set gridz " 0.000 0.050 0.000"
set xmin -1.00; set xmax 1.00
set ymin 0.01; set ymax 1.00
set zmin -1.00; set zmax 1.00
set grp_id 5
# write the matlab script
set fileID [open "parameter.m" w]
puts $fileID "normaldir = $surfacenormal"
puts $fileID "latticeconst = $lattconst"
puts $fileID "latticesize = \[ $c1 $Nx "
puts $fileID " $c2 $Ny "
puts $fileID " $c3 $Nz \]"
puts $fileID "dxyz = \[ $gridx "
puts $fileID " $gridy "
puts $fileID " $gridz \]"
close $fileID
MD++ {
element0 = Si
crystalstructure = diamond-cubic
}
MD++ latticeconst = $lattconst
MD++ latticesize = \[$c1 $Nx $c2 $Ny $c3 $Nz\]
MD++ makecrystal
MD++ input = \[1 -1 1 0.4 1 -1 1 \] fixatoms_by_position removefixedatoms
MD++ input = \[1 -1 1 -1 -0.42 -1 1 \] fixatoms_by_position removefixedatoms
MD++ input = \[ $surfacenormal $gridx $gridy $gridz $xmin $xmax $ymin $ymax $zmin $zmax 1 $grp_id \]
MD++ calmisfit2_rigidrlx
MD++ quit
} else {
puts "unknown status = $status"
MD++ quit
}
Matlab Program cal_idealstr.m
%#! /usr/local/bin/octave -qf
% Matlab script, cal_idealstr.m
% to calculate ideal tensile strength "sigma" and shear strength "tau"
clear
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% calculate ideal tensile strength along [110] direction
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
data = load(’Esurf.dat’);
d = data(:,1); % openning distance in A
gamma = data(:,2); % excessive energy per area creating 2 free surfaces (eV/A^2)
sigma = max(gamma(2:end)-gamma(1:end-1))/(d(2) - d(1)) * 160.2;
disp(sprintf(’Ideal tensile stregnth = %e(GPa)’,sigma));
figure(1)
set(gca,’fontsize’,14)
plot(d, gamma, ’.-’), grid
xlabel(’Opening Distance \it d \rm(Angstrom)’,’fontsize’,16),
ylabel(’\gamma (eV/Angstrom^2)’,’fontsize’,16)
line([-1 1.1],[gamma(end) gamma(end)],’linewidth’,2,’color’,’k’)
text(-1.3, gamma(end),’2\gamma_s’,’fontsize’,16)
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% calculate ideal shear strength on (111) along [110] direction
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
if (~exist(’parameter.m’,’file’))
error(’No parameter.m found’)
else
parameter
dx = dxyz(1,2); dy = dxyz(2,2); dz = dxyz(3,2);
vx = latticesize(1,1:3); Lx = latticeconst*norm(vx);
vy = latticesize(2,1:3); Ly = latticeconst*norm(vy);
vz = latticesize(3,1:3); Lz = latticeconst*norm(vz);
data = load(’Emisfit.out’);
Mx=max(data(:,1)); My=max(data(:,2)); Mz=max(data(:,3));
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% convert data to relaxed energy
MM=-sort(-[Mx+1,My+1,Mz+1]); % Then MM(1) > MM(2) > MM(3)
E = reshape(data(:,4),MM(1),MM(2));
Erlx = zeros(MM(1),1);
Zrlx = zeros(MM(1),1);
normalgrid = dxyz(normaldir,1):dxyz(normaldir,2):dxyz(normaldir,3);
normalfinegrid = dxyz(normaldir,1):dxyz(normaldir,2)/1000:dxyz(normaldir,3);
for i = 1:MM(1),
a = E(i,:);
if length(a)> 1
a_interp = interp1(normalgrid,a,normalfinegrid,'spline');
else
a_interp = a;
end
[Erlx(i), ind] = min(a_interp);
Zrlx(i) = normalfinegrid(ind);
end
% retrieve unrelaxed energy
Eunrlx=E(:,find(abs(normalgrid)<1e-8));
% calculate ideal shear strength
tau = max(Erlx(2:end)-Erlx(1:end-1))/(dx*Lx) * 160.2;
disp(sprintf(’Ideal shear stregnth = %e(GPa)’,tau));
figure(2); set(gca,’fontsize’,14)
plot([0:Mx]/Mx, Erlx-Erlx(1), ’b.-’,...
[0:Mx]/Mx, Eunrlx-Eunrlx(1),’r-’);
legend(’relaxed’,’unrelaxed’);
xlabel(’Sheared Distance, d/|b| where b = [1 0 1]/2’,’fontsize’,16)
ylabel(’\Gamma (eV/Angstrom^2)’,’fontsize’,16)
figure(3); set(gca,’fontsize’,14)
plot([0:Mx]/Mx,Zrlx, ’r.-’, ...
[0 1],[1 1]*dxyz(normaldir,1),’b-’,...
[0 1],[1 1]*dxyz(normaldir,3),’b-’);
xlabel(’Sheared Distance, d/|b| where b = [1 0 1]/2’,’fontsize’,16)
ylabel({’Lowest energy height,’; ’\delta/|c_2| where c_2 = [1 1 1]’},’fontsize’,16)
end
Shell script for Computing Ideal Tensile strength by VASP
The following shell script will create the POSCAR file which specifies the atomic positions for VASP. You will also need to prepare INCAR, KPOINTS, POTCAR files in the same directory for VASP to run. This script will run VASP in serial mode. You can modify it to submit VASP jobs in parallel.
In this script, the length of the first lattice vector is changed from 0.9 to 1.6 of its equilibrium length in steps of 0.01. A VASP calculation is performed for each length of the lattice vector to obtain the potential energy, which is then saved into file summary.
#!/bin/bash declare -i x=9000 while (($x <= 16000)) do a=$(echo "scale=3; $x/10000" | bc -l) cat > POSCAR << FIN Si comment line 5.389 universal scaling factor $a $a 0.0 first lattice vector -1.0 1.0 0.0 second lattice vector 0.0 0.0 1.0 third lattice vector 16 number of atoms per species selective dynamics cart direct or cart (only first letter is significant) -0.5000000000000000 0.5000000000000000 0.0000000000000000 F F F -0.2500000000000000 0.7500000000000000 0.2500000000000000 F F F -0.7500000000000000 0.7500000000000000 0.7500000000000000 F F F -0.2500000000000000 0.2500000000000000 0.7500000000000000 F F F -0.5000000000000000 1.0000000000000000 0.5000000000000000 F F F -0.2500000000000000 1.2500000000000000 0.7500000000000000 F F F 0.0000000000000000 0.0000000000000000 0.0000000000000000 F F F 0.5000000000000000 0.5000000000000000 0.0000000000000000 F F F 0.0000000000000000 0.5000000000000000 0.5000000000000000 F F F 0.2500000000000000 0.2500000000000000 0.2500000000000000 F F F 0.7500000000000000 0.7500000000000000 0.2500000000000000 F F F 0.2500000000000000 0.7500000000000000 0.7500000000000000 F F F 0.0000000000000000 1.0000000000000000 0.0000000000000000 F F F 0.0000000000000000 1.5000000000000000 0.5000000000000000 F F F 0.5000000000000000 1.0000000000000000 0.5000000000000000 F F F 0.2500000000000000 1.2500000000000000 0.2500000000000000 F F F FIN vasp E=`tail -1 OSZICAR` echo $a $E >> summary let x+=100 done
Shell script for Computing Ideal Shear strength by VASP
The following shell script will create the POSCAR file which specifies the atomic positions for VASP. You will also need to prepare INCAR, KPOINTS, POTCAR files in the same directory for VASP to run. This script will run VASP in serial mode. You can modify it to submit VASP jobs in parallel.
In this script, position of a set of atoms is displaced along the y-direction [1 0 -1] but are allowed to relax in the x-direction [1 1 1]. A VASP calculation is performed for each length of the lattice vector to obtain the potential energy, which is then saved into file summary.
#!/bin/bash
function add() {
echo "scale=17; $1 + $2" | bc -l
}
function subtract() {
echo "scale=17; $1 - $2" | bc -l
}
function div() {
echo "scale=3; $1 / $2" | bc -l
}
declare -i x=0
while (($x <= 50))
do
a=$(div $x 100)
cat > POSCAR << FIN
Si comment line
5.389 universal scaling factor
2.0 2.0 2.0 first lattice vector
1.0 0.0 -1.0 second lattice vector
-0.5 1.0 -0.5 third lattice vector
32 number of atoms per species
selective dynamics
direct direct or cart (only first letter is significant)
-0.3125000000000000 0.2500000000000000 0.0000000000000000 T F F
-0.3125000000000000 -0.2500000000000000 0.0000000000000000 T F F
-0.3125000000000000 0.0000000000000000 -0.5000000000000000 T F F
-0.2708333333333334 -0.2500000000000000 -0.3333333333333334 T F F
-0.3125000000000000 -0.5000000000000000 -0.5000000000000000 T F F
-0.1458333333333334 -0.2500000000000000 -0.3333333333333334 T F F
-0.2708333333333334 0.0000000000000000 0.1666666666666666 T F F
-0.1458333333333334 0.0000000000000000 0.1666666666666666 T F F
-0.2708333333333334 -0.5000000000000000 0.1666666666666666 T F F
-0.1041666666666667 -0.2500000000000000 0.3333333333333333 T F F
-0.1041666666666667 -0.5000000000000000 -0.1666666666666667 T F F
-0.1458333333333334 -0.5000000000000000 0.1666666666666666 T F F
0.0208333333333333 $(add -0.2500000000000000 $a) 0.3333333333333333 T F F
0.0208333333333333 $(add -0.5000000000000000 $a) -0.1666666666666667 T F F
-0.2708333333333334 0.2500000000000000 -0.3333333333333334 T F F
-0.1458333333333334 0.2500000000000000 -0.3333333333333334 T F F
-0.1041666666666667 0.2500000000000000 0.3333333333333333 T F F
0.0208333333333333 $(add 0.2500000000000000 $a) 0.3333333333333333 T F F
-0.1041666666666667 0.0000000000000000 -0.1666666666666667 T F F
0.0208333333333333 $(add 0.0000000000000000 $a) -0.1666666666666667 T F F
0.0625000000000000 $(add 0.2500000000000000 $a) 0.0000000000000000 T F F
0.0625000000000000 $(add -0.2500000000000000 $a) 0.0000000000000000 T F F
0.0625000000000000 $(add 0.0000000000000000 $a) -0.5000000000000000 T F F
0.1875000000000000 $(add 0.2500000000000000 $a) 0.0000000000000000 T F F
0.1875000000000000 $(add -0.2500000000000000 $a) 0.0000000000000000 T F F
0.1875000000000000 $(add 0.0000000000000000 $a) -0.5000000000000000 T F F
0.0625000000000000 $(add -0.5000000000000000 $a) -0.5000000000000000 T F F
0.2291666666666666 $(add -0.2500000000000000 $a) -0.3333333333333334 T F F
0.1875000000000000 $(add -0.5000000000000000 $a) -0.5000000000000000 T F F
0.2291666666666666 $(add 0.0000000000000000 $a) 0.1666666666666666 T F F
0.2291666666666666 $(add -0.5000000000000000 $a) 0.1666666666666666 T F F
0.2291666666666666 $(add 0.2500000000000000 $a) -0.3333333333333334 T F F
FIN
vasp
E=`tail -1 OSZICAR`
echo $a $E >> summary
let x+=5
done